Scalable Synthesis of High-Purity S-(-)-Nadifloxacin for Advanced Antibiotic Formulations
Scalable Synthesis of High-Purity S-(-)-Nadifloxacin for Advanced Antibiotic Formulations
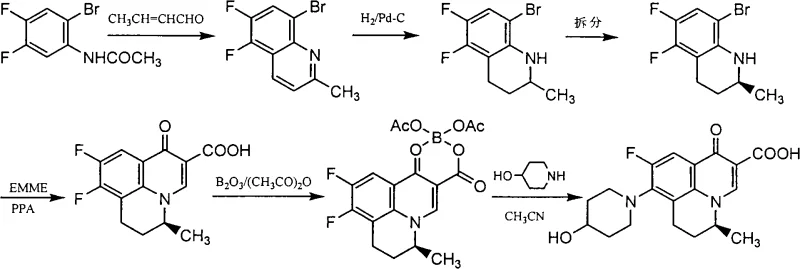
The pharmaceutical landscape for quinolone antibiotics is continuously evolving, driven by the need for higher efficacy and improved pharmacokinetic profiles. Patent CN101298452A introduces a robust and industrially viable methodology for the preparation of S-(-)-Nadifloxacin and its water-soluble salts, addressing critical limitations associated with the racemic mixture. This technology leverages a strategic chiral resolution of a key tetrahydroquinoline intermediate, ensuring high optical purity while maintaining cost-effectiveness through the use of readily available starting materials like 3,4-difluoro-acetbromanilide. For R&D directors and procurement specialists, this route represents a significant advancement, offering a pathway to produce high-purity API intermediates with superior solubility characteristics suitable for diverse dosage forms including injections and topical gels.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditional synthesis pathways for Nadifloxacin often result in a racemic mixture, containing both R and S enantiomers. Clinical data indicates that the S-(-) enantiomer possesses significantly stronger antimicrobial activity and lower toxicity compared to its R-(+) counterpart. However, conventional manufacturing frequently relies on synthesizing the racemate first, followed by difficult and yield-loss-prone separation techniques, or simply marketing the less effective mixture. Furthermore, the free acid form of Nadifloxacin exhibits extremely poor water solubility, typically less than 0.1% at room temperature. This physicochemical limitation severely restricts formulation options, largely confining the drug to suspension ointments with limited bioavailability and making the development of injectable or rapid-release oral formulations nearly impossible without complex solubilization strategies.
The Novel Approach
The innovative process disclosed in the patent fundamentally shifts the paradigm by introducing chirality early in the synthesis sequence. Instead of struggling with the final API, the method focuses on the direct resolution of the intermediate 5,6-difluoro-2-methyl-1,2,3,4-tetrahydroquinoline. By employing chiral organic acids such as L-tartrate or D-camphorsulfonic acid, the process selectively precipitates the desired S-(-) enantiomer as a salt. This intermediate is then carried forward through cyclization and condensation steps. Additionally, the patent details the formation of various water-soluble salts (e.g., sodium, potassium, arginine salts) which dramatically enhance solubility to levels exceeding 150 mg/ml. This dual approach of early chiral resolution and salt formation solves both the efficacy and formulation challenges simultaneously.
Mechanistic Insights into Chiral Resolution and Quinolone Cyclization
The core of this synthesis lies in the precise control of stereochemistry during the intermediate stage. The process begins with a Skraup quinoline synthesis using 3,4-difluoro-6-bromobenzene ethanamide and crotonic aldehyde, followed by catalytic hydrogenation using Pd-C to remove the bromine atom and saturate the ring. The resulting racemic amine is then subjected to resolution. Mechanistically, the chiral acid forms a diastereomeric salt with the amine. Due to differences in solubility and crystal lattice energy, the S-(-) salt precipitates preferentially from solvents like ethyl acetate or acetone. This solid is isolated, washed, and then neutralized with a strong base like NaOH to liberate the free chiral amine. This step is critical as it establishes the optical purity before the more complex ring-closing reactions occur, ensuring that downstream impurities do not compromise the enantiomeric excess.
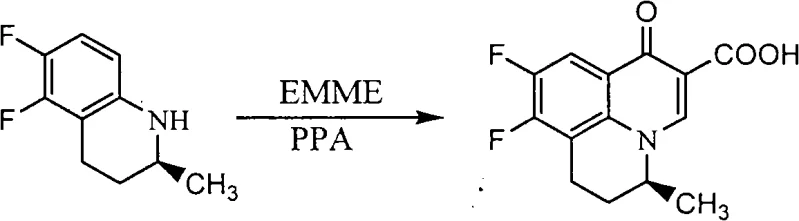
Following resolution, the chiral amine undergoes cyclization with ethoxymethylene malonate (EMME) in the presence of polyphosphoric acid (PPA) at elevated temperatures around 130°C. This step constructs the quinolone backbone. To facilitate the subsequent nucleophilic substitution with 4-hydroxypiperidine, the carboxylic acid group at the C-3 position is protected. This is achieved using a boron complex formed from boric anhydride and acetic anhydride. This protection strategy prevents side reactions and improves the regioselectivity of the piperidine condensation. Finally, alkaline hydrolysis removes the boron protecting group and hydrolyzes the ester, yielding the final S-(-)-Nadifloxacin. The entire sequence is designed to minimize impurity generation, particularly avoiding the formation of decarboxylated byproducts common in quinolone synthesis.
How to Synthesize S-(-)-Nadifloxacin Efficiently
The synthesis of this high-value antibiotic intermediate requires strict adherence to reaction parameters to ensure consistent optical purity and yield. The process integrates classical organic transformations with modern chiral separation techniques, making it highly adaptable for commercial scale-up. Operators must pay close attention to the stoichiometry of the resolving agent and the temperature control during the PPA cyclization step to prevent degradation. The following guide outlines the standardized operational framework derived from the patent examples, providing a clear roadmap for laboratory and pilot plant execution.
- Perform Skraup quinoline synthesis followed by catalytic hydrogenation to obtain the racemic tetrahydroquinoline intermediate, then resolve using L-tartrate or similar chiral acids to isolate the S-(-) enantiomer.
- Execute ring closure using ethoxymethylene malonate (EMME) in polyphosphoric acid (PPA) to form the quinolone core, followed by carboxyl protection with boric anhydride.
- Condense the protected intermediate with 4-hydroxypiperidine in acetonitrile, followed by alkaline hydrolysis and deprotection to yield the final S-(-)-Nadifloxacin or its water-soluble salts.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the economic and logistical benefits of this patented route are substantial. The reliance on commodity chemicals like 3,4-difluoro-acetbromanilide and crotonic aldehyde ensures a stable and cost-effective raw material supply chain, insulating production from the volatility often associated with exotic chiral starting materials. Furthermore, the ability to recycle the chiral resolving agents significantly reduces the net consumption of these auxiliaries, directly contributing to cost reduction in antibiotic manufacturing without compromising quality. The process avoids the use of expensive transition metal catalysts for asymmetric synthesis, relying instead on stoichiometric resolution which is often more predictable and easier to validate in a GMP environment.
- Cost Reduction in Manufacturing: The elimination of complex asymmetric catalysis and the implementation of a recyclable resolution strategy lead to significant operational savings. By recovering and reusing chiral acids like L-tartrate, the effective cost per kilogram of the chiral intermediate is drastically lowered. Additionally, the high solubility of the final salt forms reduces the need for expensive solubilizing excipients in the final drug product, further optimizing the overall cost structure for the finished dosage form.
- Enhanced Supply Chain Reliability: The synthetic route utilizes robust, well-understood chemical transformations such as hydrogenation and acid-base extraction, which are easily scalable from kilograms to multi-ton quantities. This simplicity reduces the risk of batch failures and supply interruptions. The availability of multiple salt forms (sodium, potassium, amino acid salts) provides formulation flexibility, allowing supply chain planners to adapt to different regional regulatory requirements or manufacturing capabilities without altering the core API synthesis.
- Scalability and Environmental Compliance: The process is designed with industrialization in mind, utilizing solvents like ethyl acetate and acetonitrile which are manageable in standard waste treatment facilities. The avoidance of heavy metal catalysts simplifies the purification process and reduces the environmental burden associated with metal residue disposal. The high yields reported in the patent examples, particularly in the hydrogenation and protection steps, indicate a material-efficient process that minimizes waste generation and maximizes throughput.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of S-(-)-Nadifloxacin. These insights are derived directly from the experimental data and technical specifications outlined in the patent documentation, providing clarity on the feasibility and advantages of this specific manufacturing route for potential partners and licensees.
Q: How does the chiral resolution step impact the optical purity of the final API?
A: The process utilizes chiral organic acids like L-tartrate or D-camphorsulfonic acid to selectively salify the S-(-)-tetrahydroquinoline intermediate. This resolution step ensures high optical purity before the final cyclization, preventing the formation of the less active R-enantiomer and reducing downstream purification burdens.
Q: What are the solubility advantages of the S-(-)-Nadifloxacin salts compared to the free acid?
A: The free acid form of Nadifloxacin has very poor water solubility (less than 0.1%). By forming salts with pharmaceutically acceptable bases such as sodium hydroxide, arginine, or lysine, the solubility increases drastically to over 150 mg/ml, enabling the development of injectable formulations and improving bioavailability.
Q: Is the resolving agent used in the synthesis recyclable for cost efficiency?
A: Yes, the patent highlights that the chiral resolving agents, such as tartaric acid derivatives, can be recovered and recycled after the neutralization and extraction steps. This significantly lowers the raw material cost per kilogram of the final chiral intermediate compared to asymmetric synthesis routes requiring expensive chiral catalysts.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable S-(-)-Nadifloxacin Supplier
At NINGBO INNO PHARMCHEM, we understand the critical importance of purity and consistency in the production of chiral antibiotics. Our technical team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the intricate chiral resolution and cyclization steps described in this patent are executed with precision. We operate stringent purity specifications and maintain rigorous QC labs equipped to analyze enantiomeric excess and trace impurities, guaranteeing that every batch of S-(-)-Nadifloxacin meets the highest international standards for pharmaceutical intermediates and APIs.
We invite global pharmaceutical partners to collaborate with us on optimizing their supply chains for next-generation quinolones. Our engineering team is ready to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements. Please contact our technical procurement team to request specific COA data, route feasibility assessments, and samples for your formulation development projects. Let us help you bring safer, more effective antibiotic therapies to market faster.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →