Scalable Synthesis of 6-Chloro-3-Methyl Uracil for High-Purity Alogliptin Manufacturing
Scalable Synthesis of 6-Chloro-3-Methyl Uracil for High-Purity Alogliptin Manufacturing
The pharmaceutical landscape for Type 2 diabetes treatments continues to evolve, driving intense demand for high-quality intermediates such as 6-chloro-3-methyl uracil, a critical precursor for the DPP-4 inhibitor Alogliptin. Patent CN108586360B introduces a robust and environmentally superior preparation method that addresses longstanding inefficiencies in heterocyclic synthesis. By shifting away from hazardous alkylation agents toward a controlled alkaline cyclization strategy, this technology offers a compelling value proposition for global supply chains seeking reliability and compliance. The process leverages the nucleophilic potential of N-methylurea reacting with dimethyl malonate under precise thermal regulation, ensuring minimal byproduct formation. For R&D directors and procurement specialists, understanding the mechanistic advantages of this route is essential for securing a stable supply of this key pharmaceutical building block.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of 6-chloro-3-methyl uracil has been plagued by significant technical and economic drawbacks that hinder efficient commercial scale-up of complex pharmaceutical intermediates. Traditional pathways often rely on the direct alkylation of 6-chlorouracil using methyl iodide, a reagent known for its severe toxicity, volatility, and high cost, which imposes heavy burdens on safety protocols and waste management systems. Alternatively, cyclization methods utilizing free malonic acid frequently suffer from poor reaction kinetics and low conversion rates, with patent literature indicating overall yields often falling below 60%. These inefficiencies result in substantial material loss, increased solvent consumption for purification, and a final product quality that may struggle to meet the rigorous impurity specifications required for modern API manufacturing. Furthermore, the use of strong acids or harsh conditions in older methods can lead to equipment corrosion and unpredictable exothermic events, creating bottlenecks in continuous production environments.
The Novel Approach
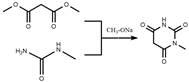
In stark contrast, the methodology disclosed in CN108586360B employs a sophisticated cyclization strategy using dimethyl malonate and N-methylurea mediated by sodium methoxide, effectively bypassing the need for toxic methylating agents. This novel approach capitalizes on the in situ generation of reactive enolates under mild alkaline conditions, promoting a clean ring-closure reaction that inherently suppresses the formation of polymeric byproducts. The process is designed with safety and scalability in mind, utilizing methanol as a benign solvent system that simplifies downstream recovery and recycling operations. By carefully controlling the addition rate of dimethyl malonate and maintaining temperatures between 40-50°C during the initial phase, the reaction avoids the rapid hydrolysis that typically plagues ester-based syntheses in aqueous or acidic media. This results in a significantly streamlined workflow where the intermediate 1-methyl barbituric acid precipitates with high purity, reducing the load on subsequent purification steps and enhancing the overall economic viability of the manufacturing process.
Mechanistic Insights into Alkaline Cyclization and Chlorination
The core of this synthetic breakthrough lies in the precise manipulation of reaction equilibria during the cyclization step, where sodium methoxide acts not merely as a base but as a crucial promoter of nucleophilic attack. In this mechanism, the methoxide ion deprotonates the N-methylurea, generating a highly reactive nitrogen nucleophile that attacks the carbonyl carbon of the dimethyl malonate ester. Unlike acid-catalyzed routes where protonation can lead to unwanted ester hydrolysis before cyclization occurs, this alkaline environment stabilizes the tetrahedral intermediate, facilitating the elimination of methanol and the subsequent closure of the pyrimidine ring. The careful dropwise addition of the diester ensures that the local concentration of reactants remains optimal, preventing intermolecular side reactions that could lead to oligomerization. This kinetic control is vital for achieving the high purity profiles demanded by regulatory bodies, as it minimizes the generation of structural analogs that are difficult to separate in later stages.
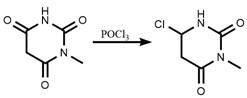
Following the isolation of the 1-methyl barbituric acid intermediate, the process transitions to a chlorination step utilizing phosphorus oxychloride (POCl3), a standard yet highly effective reagent for introducing chlorine atoms at the C-6 position of the uracil ring. The mechanism involves the activation of the carbonyl oxygen by the phosphorus center, creating a good leaving group that is subsequently displaced by a chloride ion. To manage the exothermic nature of this reaction and ensure safety, the protocol specifies a controlled temperature ramp to 70-75°C, allowing for complete conversion without thermal degradation of the sensitive heterocyclic core. Crucially, the workup procedure involves a neutralization step where residual sodium methoxide and basic byproducts are quenched with hydrochloric acid, generating harmless sodium chloride and methanol which are easily removed. This thoughtful integration of reaction and workup chemistry ensures that the final 6-chloro-3-methyl uracil is obtained with minimal inorganic contamination, ready for the next stage of API synthesis.
How to Synthesize 6-Chloro-3-Methyl Uracil Efficiently
Implementing this synthesis requires strict adherence to the thermal and stoichiometric parameters outlined in the patent to maximize yield and safety. The process begins with the preparation of a methanolic solution of sodium methoxide, into which N-methylurea is dissolved under inert atmosphere to prevent moisture ingress. Dimethyl malonate is then introduced slowly over a period of 1 to 2 hours while maintaining the reaction mixture at 40-50°C, followed by a reflux period to drive the cyclization to completion. Once the 1-methyl barbituric acid precipitates upon cooling, it is isolated via filtration and subjected to an acid wash to remove residual bases before drying. The dried solid is then suspended in phosphorus oxychloride, and water is added dropwise at low temperature to initiate the chlorination, followed by heating to 70-75°C. Detailed standardized operating procedures for each unit operation, including specific mass ratios and stirring speeds, are critical for reproducibility.
- Cyclize dimethyl malonate with N-methylurea using sodium methoxide in methanol at 40-50°C to form 1-methyl barbituric acid.
- Purify the intermediate by acidification with hydrochloric acid to remove sodium salts and isolate the solid crystals.
- React the dried 1-methyl barbituric acid with phosphorus oxychloride (POCl3) at 70-75°C to effect chlorination and obtain the final product.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this patented route translates into tangible strategic benefits regarding cost stability and operational continuity. By eliminating the reliance on methyl iodide, a volatile and regulated substance subject to fluctuating market prices and strict transportation controls, manufacturers can secure a more predictable raw material supply chain. The substitution of expensive and hazardous reagents with commodity chemicals like dimethyl malonate and sodium methoxide drastically simplifies logistics and reduces the overhead associated with hazardous waste disposal. Furthermore, the improved reaction efficiency means that less raw material is required per kilogram of finished product, directly contributing to substantial cost savings in high-purity pharmaceutical intermediate manufacturing. The simplified purification process, which relies on crystallization rather than complex chromatography, also reduces solvent consumption and processing time, enhancing the overall throughput of the production facility.
- Cost Reduction in Manufacturing: The elimination of methyl iodide removes a significant cost driver associated with both raw material acquisition and the specialized containment systems required for its handling. Additionally, the higher yield profile of this cyclization method means that the effective cost per unit of output is lowered, as less feedstock is wasted in side reactions or lost during extensive purification cycles. The use of methanol as a primary solvent further aids in cost optimization, as it is inexpensive, readily available in bulk, and easily recovered for reuse, minimizing the environmental levy and utility costs associated with solvent incineration.
- Enhanced Supply Chain Reliability: Sourcing dimethyl malonate and urea derivatives is far less risky than procuring specialized alkylating agents, as these are produced by a wide range of global chemical suppliers, ensuring redundancy in the supply base. The robustness of the reaction conditions, which tolerate minor variations in temperature and addition rates without catastrophic failure, makes the process highly reliable for contract manufacturing organizations (CMOs) managing tight delivery schedules. This resilience reduces the likelihood of batch failures or delays, ensuring a consistent flow of material to downstream API synthesis sites and preventing costly production stoppages.
- Scalability and Environmental Compliance: The process is inherently designed for scale-up, utilizing standard reactor configurations and avoiding high-pressure or cryogenic conditions that limit batch size. From an environmental perspective, the generation of benign byproducts like sodium chloride and methanol simplifies effluent treatment, helping facilities meet increasingly stringent discharge regulations without investing in exotic remediation technologies. This alignment with green chemistry principles not only mitigates regulatory risk but also enhances the corporate sustainability profile of the manufacturing entity, a key factor for partnerships with major multinational pharmaceutical companies.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis route, derived directly from the experimental data and beneficial effects described in the patent documentation. Understanding these nuances is critical for technical teams evaluating the feasibility of technology transfer or process validation. The answers reflect the specific advantages of the alkaline cyclization method over traditional acid-catalyzed or alkylation-based approaches, focusing on yield, purity, and safety metrics.
Q: Why is this synthesis route preferred over traditional methyl iodide methods?
A: Traditional methods often utilize methyl iodide, which is highly toxic, environmentally hazardous, and expensive. This patented route utilizes dimethyl malonate and sodium methoxide, significantly reducing environmental impact and operational safety risks while improving overall yield.
Q: What is the expected purity profile for this intermediate?
A: The process described in patent CN108586360B demonstrates the ability to achieve purity levels exceeding 98% through optimized recrystallization steps, making it suitable for stringent pharmaceutical applications like Alogliptin synthesis.
Q: Is this process suitable for large-scale industrial production?
A: Yes, the reaction conditions (40-75°C) are moderate and utilize common solvents like methanol. The workup involves simple filtration and centrifugation, avoiding complex chromatographic separations, which facilitates easy scale-up from pilot to commercial tonnage.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 6-Chloro-3-Methyl Uracil Supplier
At NINGBO INNO PHARMCHEM, we recognize that the successful commercialization of diabetes therapeutics depends on the uninterrupted supply of high-quality intermediates like 6-chloro-3-methyl uracil. As a leading CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project needs are met with precision and speed. Our facilities are equipped with rigorous QC labs capable of verifying stringent purity specifications, guaranteeing that every batch meets the exacting standards required for GMP API synthesis. We combine deep technical expertise with a commitment to operational excellence, making us the ideal partner for navigating the complexities of modern pharmaceutical supply chains.
We invite you to engage with our technical procurement team to discuss how this advanced synthesis route can be integrated into your supply strategy. By requesting a Customized Cost-Saving Analysis, you can gain deeper insights into the economic benefits of switching to this greener, more efficient process. We encourage potential partners to contact us directly to obtain specific COA data and route feasibility assessments tailored to your specific volume requirements and quality targets.