Advanced Synthetic Route for Cediranib Intermediates Enhancing Commercial Scalability and Purity
The pharmaceutical industry continuously seeks robust synthetic pathways for potent oncology agents, and the synthesis of Cediranib (AZD2171), a pan-VEGF receptor tyrosine kinase inhibitor, represents a critical area of development. Patent CN102603718A discloses a refined preparation method that significantly addresses the limitations of prior art, offering a pathway characterized by mild reaction conditions, enhanced yields, and superior scalability. This technical insight report analyzes the proprietary methodology detailed in the patent, highlighting its potential for industrial application. By utilizing trifluoronitrobenzene and methyl vanillate as primary starting materials, the process navigates through strategic acetylation, substitution, cyclization, and protection steps to construct the complex molecular architecture required for this anticancer therapeutic. For procurement leaders and R&D directors, understanding these mechanistic nuances is vital for securing a reliable API intermediate supplier capable of delivering consistent quality.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historical synthetic routes for Cediranib intermediates have been plagued by significant operational challenges that hinder efficient cost reduction in pharmaceutical manufacturing. Prior patents, such as WO2000047212, relied on trimethyl orthoformate for ketal protection, necessitating multiple protection and deprotection steps that complicated purification and lowered overall throughput. Other methods, described in WO2004009542, utilized sodium methylate nucleophilic substitution followed by high-temperature reactions at 180°C to introduce hydroxyl groups, resulting in excessive energy consumption and safety risks. Furthermore, traditional reduction methods often employed zinc or iron powder in acidic media, generating substantial amounts of solid metal sludge that created severe environmental disposal issues and complicated downstream processing. The use of concentrated sulfuric acid in hydrolysis steps frequently led to carbonization of the product, introducing difficult-to-remove impurities that compromised the purity profile essential for high-purity Cediranib intermediates.
The Novel Approach
The methodology outlined in CN102603718A introduces a paradigm shift by optimizing reaction conditions to mitigate these historical pain points. Instead of harsh sulfuric acid hydrolysis, the novel route employs a mixture of concentrated hydrochloric acid and glacial acetic acid at moderate temperatures (60-120°C), effectively preventing carbonization while maintaining high conversion rates. A standout innovation is the replacement of metal powder reductions with sodium dithionite (V-Brite B) in the presence of potassium carbonate, which facilitates clean reductive cyclization at ambient to mild temperatures (0-40°C). Additionally, the cyclization step to form the quinazoline core utilizes formamidine acetate rather than formamide, allowing the reaction to proceed at significantly lower temperatures (60-100°C versus 190°C). These modifications collectively streamline the workflow, reduce waste generation, and enhance the feasibility of commercial scale-up of complex oncology intermediates.
Mechanistic Insights into Sodium Dithionite-Mediated Reductive Cyclization
The construction of the indole core is a pivotal moment in the synthesis, and the patent details a sophisticated reductive cyclization mechanism. In Step 4, the intermediate 1-(3-(benzyloxy)-2-fluoro-6-nitrophenyl)propan-2-one undergoes transformation into 5-(benzyloxy)-4-fluoro-2-methyl-1H-indole. This is achieved using sodium dithionite as the reducing agent in an ethanol-water solvent system catalyzed by potassium carbonate. Unlike catalytic hydrogenation which might require high pressure or specialized equipment, or metal reductions which produce sludge, sodium dithionite offers a homogeneous reduction environment. The mechanism likely involves the generation of sulfoxylate radicals that reduce the nitro group to an amine in situ, which immediately attacks the adjacent ketone carbonyl to close the ring. This tandem reduction-cyclization sequence is highly efficient, reportedly achieving yields as high as 98% in experimental embodiments.
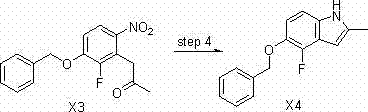
Impurity control is rigorously managed throughout this sequence. The patent specifies precise solvent ratios during workup, such as using a methylene chloride to petroleum ether mixture (1:0.5 to 1:2) to precipitate solid products directly from the reaction mixture. This crystallization-induced purification strategy minimizes the need for column chromatography, which is often a bottleneck in large-scale production. Furthermore, the avoidance of high-temperature acidic conditions prevents the formation of polymeric tars and carbonized byproducts common in older methods. The subsequent deprotection step utilizes 10% Pd/C under atmospheric hydrogenation, a standard yet highly effective method for removing the benzyl protecting group to reveal the crucial phenolic hydroxyl moiety required for final coupling. This attention to detail in mechanistic execution ensures that the final high-purity Cediranib intermediates meet stringent regulatory specifications.
How to Synthesize Cediranib Intermediates Efficiently
The synthesis of Cediranib involves a convergent strategy where two major fragments—the fluoro-methyl-indole segment and the methoxy-quinazoline segment—are prepared separately and then coupled. The process begins with the nucleophilic aromatic substitution of trifluoronitrobenzene with methyl acetoacetate, followed by decarboxylation to establish the side chain. Parallel to this, methyl vanillate is protected, nitrated, reduced, and cyclized to form the quinazoline core. The convergence occurs via a nucleophilic substitution between the chloro-quinazoline and the indole phenol.
- Condense methyl acetoacetate with trifluoronitrobenzene using sodium ethylate catalyst in THF at 0-40°C to form the nitrophenyl acetoacetate derivative.
- Perform decarboxylation and hydrolysis using hydrochloric acid and glacial acetic acid at 60-120°C to obtain the propyl ketone intermediate.
- Execute reductive cyclization using sodium dithionite and potassium carbonate in ethanol/water to construct the fluoro-methyl-indole core structure.
Commercial Advantages for Procurement and Supply Chain Teams
For supply chain stakeholders, the transition to this optimized synthetic route offers tangible logistical and economic benefits beyond simple yield metrics. The elimination of hazardous reagents like concentrated sulfuric acid and bulk metal powders significantly reduces the burden on waste treatment facilities, thereby lowering the total cost of ownership for the manufacturing process. The reliance on readily available commodity chemicals such as vanillin derivatives and trifluoronitrobenzene ensures a stable supply chain, mitigating the risk of raw material shortages that can plague specialty syntheses. Moreover, the milder reaction temperatures reduce energy consumption and extend the lifespan of reactor vessels, contributing to long-term operational sustainability.
- Cost Reduction in Manufacturing: The substitution of expensive and wasteful metal powder reductions with sodium dithionite eliminates the costly disposal of heavy metal sludge. Additionally, the use of formamidine acetate allows for lower temperature cyclization, reducing energy costs associated with heating reactors to extreme temperatures. These process intensifications lead to substantial cost savings without compromising product quality.
- Enhanced Supply Chain Reliability: By utilizing common starting materials and avoiding exotic catalysts that may have long lead times, the manufacturing timeline becomes more predictable. The robust nature of the reaction conditions means that batch failures due to sensitive parameters are minimized, ensuring a consistent flow of materials to downstream formulation teams and reducing lead time for high-purity Cediranib intermediates.
- Scalability and Environmental Compliance: The process is designed with industrial scalability in mind. The avoidance of carbonization and the implementation of crystallization-based purifications make the transition from pilot plant to multi-ton production seamless. Furthermore, the greener profile of the synthesis, characterized by reduced hazardous waste, aligns with modern environmental compliance standards, facilitating smoother regulatory approvals.
Frequently Asked Questions (FAQ)
The following questions address common technical inquiries regarding the implementation of this synthetic route. These insights are derived directly from the experimental data and comparative analysis provided in the patent documentation, aiming to clarify the operational advantages for potential manufacturing partners.
Q: How does this synthesis method improve upon traditional Cediranib production?
A: This method replaces harsh conditions like 180°C reactions and sulfuric acid hydrolysis with milder temperatures (0-100°C) and avoids carbonization. It also substitutes zinc/iron powder reduction with sodium dithionite, eliminating heavy metal sludge and simplifying purification.
Q: What are the key yield improvements observed in this patent?
A: The patent reports significant yield increases for key intermediates, such as the 4-fluoro-2-methyl-1H-indol-5-ol segment reaching up to 87% after deprotection, and the quinazoline segment achieving yields around 96% during chlorination, contributing to an overall improved process efficiency.
Q: Is this process suitable for large-scale commercial manufacturing?
A: Yes, the process utilizes readily available raw materials like vanillin derivatives and trifluoronitrobenzene. The avoidance of extreme temperatures and difficult-to-handle metal powders makes it highly amenable to scale-up from kilogram to multi-ton production levels.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Cediranib Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of efficient synthetic routes in the competitive landscape of oncology drug development. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the theoretical benefits of patents like CN102603718A are realized in practical, GMP-compliant manufacturing. We maintain stringent purity specifications and operate rigorous QC labs to guarantee that every batch of Cediranib intermediate meets the exacting standards required for clinical and commercial applications. Our commitment to technical excellence allows us to navigate complex chemistries with precision and reliability.
We invite global pharmaceutical partners to collaborate with us to leverage these advanced manufacturing capabilities. By engaging with our technical procurement team, you can request a Customized Cost-Saving Analysis tailored to your specific volume requirements. We encourage you to reach out for specific COA data and route feasibility assessments to determine how our optimized processes can enhance your supply chain resilience and drive value for your organization.