Advanced Synthesis of HDMS Lamivudine Intermediate for Commercial Scale Production
Advanced Synthesis of HDMS Lamivudine Intermediate for Commercial Scale Production
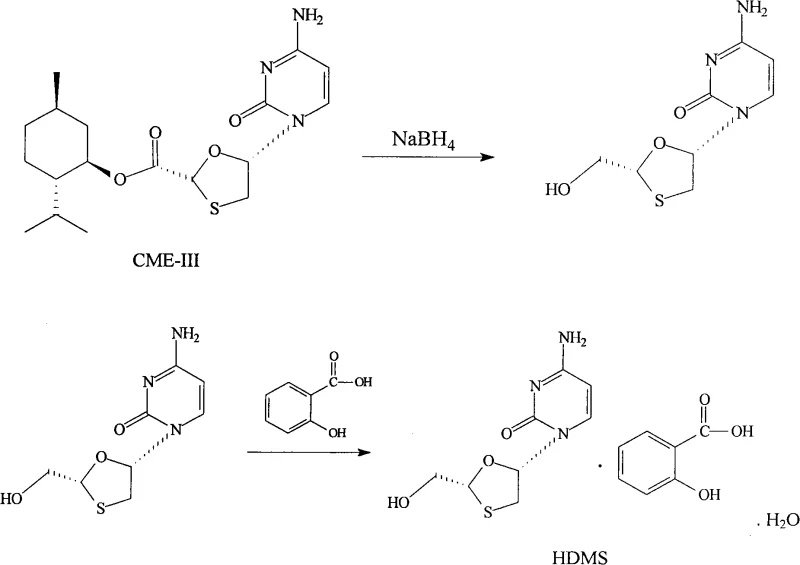
The global demand for high-purity antiviral agents continues to surge, driven by the persistent need for effective treatments against Hepatitis B and HIV. At the heart of this therapeutic landscape lies Lamivudine, a nucleoside analogue reverse transcriptase inhibitor whose efficacy is heavily dependent on the quality of its synthetic precursors. A pivotal advancement in this domain is documented in patent CN101830893A, which discloses a robust and economically viable synthesis and preparation process for the Lamivudine intermediate known as HDMS (salicylate salt). This technical breakthrough addresses long-standing challenges in the pharmaceutical industry, specifically focusing on the optimization of the glyoxylic acid menthol ester (CME-I) preparation. By shifting away from hazardous oxidants and complex purification techniques, this methodology offers a streamlined pathway that enhances both yield and operational safety. For procurement leaders and R&D directors seeking a reliable pharmaceutical intermediate supplier, understanding the nuances of this patented route is essential for securing a stable supply chain for next-generation antiviral formulations.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the industrial synthesis of Lamivudine intermediates has been plagued by significant technical bottlenecks that hinder efficient commercialization. Traditional routes, such as those described in US Patent 5047407, often rely on the reaction of 2-bromo-diethoxyethane with thiobenzoic acid, followed by hydrolysis and coupling with benzoyl oxyacetaldehyde. These legacy processes are characterized by low reaction yields and the necessity for cumbersome purification steps, including column chromatography, to separate cis and trans isomers. Furthermore, alternative methods utilizing L-tartrate and sodium periodate oxidation introduce severe safety hazards due to the use of strong oxidizing agents, which complicate waste treatment and increase the risk of runaway reactions. The reliance on such harsh reagents not only escalates production costs but also creates substantial environmental liabilities, making these conventional approaches increasingly untenable for modern, green chemistry-compliant manufacturing facilities.
The Novel Approach
In stark contrast, the novel approach outlined in the referenced patent introduces a paradigm shift by utilizing L-menthol and glyoxylic acid as primary starting materials under mild acidic catalysis. This strategy circumvents the need for dangerous oxidants and eliminates the requirement for preparative chromatography, which is a major cost driver in fine chemical synthesis. The process leverages a unique purification mechanism involving sodium bisulfite adduct formation, allowing for the selective isolation of the desired ester intermediate with high fidelity. By operating primarily in aqueous or cyclohexane-based systems at moderate temperatures ranging between 20°C and 30°C, the new method drastically reduces energy consumption and thermal risks. This innovation represents a significant leap forward in cost reduction in API manufacturing, offering a scalable solution that aligns perfectly with the rigorous quality standards demanded by international regulatory bodies.
Mechanistic Insights into Esterification and Bisulfite Purification
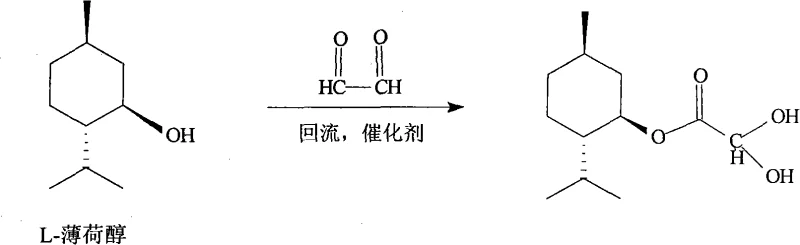
The core of this technological advancement lies in the meticulous control of the esterification reaction between L-menthol and glyoxylic acid. Under the catalysis of concentrated sulfuric acid in a cyclohexane solvent system, the reaction proceeds via a reflux mechanism that effectively removes water to drive the equilibrium towards product formation. The use of cyclohexane is particularly strategic; it serves not only as a reaction medium but also as an extraction solvent that facilitates the separation of organic and aqueous phases in subsequent steps. Crucially, the reaction endpoint is monitored using gas chromatography, ensuring that the conversion is optimized before proceeding to the purification stage. This level of process analytical technology (PAT) integration is vital for maintaining batch-to-batch consistency, a key metric for any high-purity pharmaceutical intermediate intended for clinical use.
Following the initial esterification, the crude organic layer undergoes a sophisticated purification sequence involving sodium bisulfite. At temperatures maintained strictly between 20°C and 30°C, the glyoxylic acid menthol ester reacts with sodium bisulfite to form a water-soluble adduct, leaving behind non-polar impurities in the organic phase. This step is critical for impurity control, as it effectively scrubs the product stream of unreacted starting materials and side products. Subsequent treatment with formaldehyde regenerates the free ester from the bisulfite adduct, precipitating it as a high-purity solid. This chemical switching mechanism ensures that the final CME-I intermediate meets stringent purity specifications, with impurities such as carboxylic acid controlled to levels below 0.5%. Such precise control over the impurity profile is indispensable for preventing downstream contamination in the synthesis of the final active pharmaceutical ingredient.
How to Synthesize Glyoxylic Acid Menthol Ester Efficiently
The synthesis of the key intermediate CME-I is the foundational step that determines the overall success of the HDMS production line. The patented procedure outlines a clear, step-by-step protocol that begins with the charging of cyclohexane, L-menthol, and glyoxylic acid into a reactor equipped with a Dean-Stark trap for water removal. The reaction is initiated by the addition of concentrated sulfuric acid and heated to reflux for approximately six hours. Upon completion, confirmed by GC analysis showing a specific ratio of product to starting material, the mixture is cooled and washed. The subsequent formation of the bisulfite adduct and its regeneration with formaldehyde are conducted under strictly controlled thermal conditions to prevent decomposition. For a comprehensive breakdown of the exact stoichiometric ratios, stirring speeds, and drying parameters required for GMP-compliant production, please refer to the standardized synthesis guide provided below.
- Perform reflux reaction of L-menthol and glyoxylic acid in cyclohexane with concentrated sulfuric acid catalysis for 6 hours.
- Cool the reaction mixture, wash with water, and react the organic layer with sodium bisulfite solution at 20-30°C to form the adduct.
- React the aqueous layer with formaldehyde at 20-30°C, filter, wash, and dry to obtain pure Glyoxylic acid menthol ester.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the transition to this novel synthesis route offers compelling economic and logistical benefits that extend far beyond simple yield improvements. The elimination of column chromatography, a notoriously slow and solvent-intensive purification method, translates directly into reduced cycle times and lower solvent procurement costs. Furthermore, the substitution of hazardous oxidants like sodium periodate with benign reagents such as sodium bisulfite significantly lowers the barrier for regulatory approval regarding environmental, health, and safety (EHS) compliance. This shift not only mitigates the risk of production stoppages due to safety incidents but also simplifies the waste treatment process, leading to substantial operational expenditure (OpEx) savings. By adopting this methodology, organizations can achieve a more resilient supply chain capable of withstanding market volatility and raw material fluctuations.
- Cost Reduction in Manufacturing: The economic impact of this process is profound, primarily driven by the simplification of the purification workflow. By removing the need for expensive silica gel columns and the associated large volumes of elution solvents, the direct material costs are significantly curtailed. Additionally, the high recovery rate of the cyclohexane solvent allows for closed-loop recycling, further diminishing the net consumption of raw materials. The use of readily available and inexpensive reagents like L-menthol and glyoxylic acid, combined with the high yield of the esterification step (reported around 85%), ensures a highly favorable cost-of-goods-sold (COGS) structure. These factors collectively contribute to a drastic reduction in the overall manufacturing cost of the HDMS intermediate.
- Enhanced Supply Chain Reliability: From a logistics perspective, the reliance on commodity chemicals rather than specialized, hard-to-source reagents enhances supply security. L-menthol and glyoxylic acid are produced at a global scale, reducing the risk of single-source bottlenecks that often plague complex pharmaceutical syntheses. The robustness of the reaction conditions, which tolerate minor variations in temperature and mixing without compromising product quality, adds another layer of reliability. This operational flexibility ensures that production schedules can be maintained consistently, even in the face of minor equipment variances or utility fluctuations, thereby guaranteeing on-time delivery for downstream API manufacturers.
- Scalability and Environmental Compliance: The process is inherently designed for scale-up, utilizing standard unit operations such as reflux, liquid-liquid extraction, and filtration that are easily transferable from pilot plants to multi-ton reactors. The absence of exotic catalysts or extreme pressure requirements means that existing infrastructure can often be utilized without major capital investment. Moreover, the generation of aqueous waste streams containing biodegradable salts aligns with modern green chemistry principles, facilitating easier permitting and disposal. This environmental compatibility is increasingly becoming a prerequisite for partnerships with top-tier pharmaceutical companies committed to sustainability goals.
Frequently Asked Questions (FAQ)
To further clarify the technical and commercial implications of this synthesis technology, we have compiled a set of frequently asked questions based on the patent data and industry standards. These inquiries address critical aspects regarding purity specifications, process safety, and scalability, providing decision-makers with the necessary insights to evaluate this technology for their own supply chains. Understanding these details is crucial for assessing the feasibility of integrating this intermediate into your current manufacturing portfolio. The following answers are derived directly from the technical disclosures and experimental data provided in the source documentation.
Q: What are the critical purity specifications for HDMS intermediate?
A: According to the patent data, the product should have an HPLC purity of not less than 80.0%, with specific limits on impurities like Whitfield's ointment (≤20.0%) and carboxylic acid (≤0.5%). The sum of HDMS and Whitfield's ointment must be ≥98.5%.
Q: How does this process improve upon conventional synthesis methods?
A: This method eliminates the need for complex column chromatography and harsh oxidants like sodium periodate. It utilizes milder reaction conditions (20-30°C for key steps) and safer solvents like cyclohexane, significantly improving operational safety and yield stability.
Q: Is this synthesis route suitable for large-scale industrial production?
A: Yes, the process is designed for scalability. It uses common solvents that are easy to recover, avoids expensive transition metal catalysts, and employs gas chromatography for precise endpoint monitoring, ensuring consistent quality from pilot to commercial scale.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable HDMS Supplier
As the pharmaceutical landscape evolves, the need for partners who can bridge the gap between innovative patent chemistry and commercial reality has never been greater. NINGBO INNO PHARMCHEM stands at the forefront of this transition, leveraging deep expertise in process chemistry to deliver high-value intermediates like HDMS with unmatched consistency. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the theoretical benefits of the patented route are fully realized in practice. We operate state-of-the-art rigorous QC labs that enforce stringent purity specifications, guaranteeing that every batch of HDMS meets the exacting standards required for antiviral drug synthesis. Our commitment to quality assurance ensures that our clients receive materials that facilitate smooth downstream processing and regulatory filing.
We invite global pharmaceutical partners to engage with us to explore how this advanced synthesis route can optimize your supply chain and reduce overall production costs. Our technical procurement team is ready to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements and quality targets. We encourage you to contact us directly to request specific COA data and route feasibility assessments, allowing you to make informed decisions based on hard data and proven performance. By collaborating with NINGBO INNO PHARMCHEM, you secure not just a supplier, but a strategic partner dedicated to the success of your antiviral drug development programs.