Scalable Synthesis of Chiral Hydroxy Methoxy Butyl Carbamate for Advanced Pharmaceutical Manufacturing
Introduction to Advanced Chiral Intermediate Synthesis
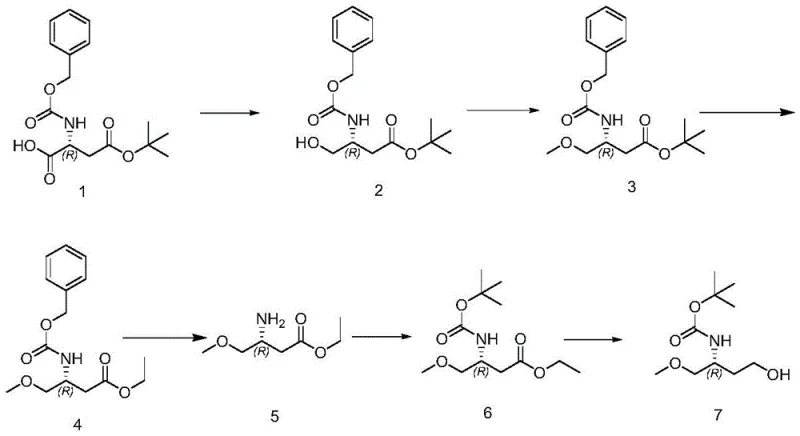
The pharmaceutical industry continuously demands high-purity chiral building blocks that can be manufactured reliably at scale. Patent CN110498750B introduces a robust synthetic methodology for producing (R)-4-hydroxy-1-methoxybut-2-yl carbamate tert-butyl ester, a critical intermediate in the construction of complex bioactive molecules. This patent outlines a six-step sequence that transforms a readily available chiral acid derivative into a functionalized amino alcohol precursor with exceptional control over stereochemistry. The process is characterized by its reliance on standard organic transformations such as mixed anhydride reduction, etherification, and catalytic hydrogenation, all optimized to maximize yield while minimizing impurity formation. For R&D directors and process chemists, this route represents a significant advancement over older methodologies that often suffered from poor selectivity or required hazardous reagents. By leveraging mild reaction conditions and commercially accessible catalysts, this technology offers a viable pathway for the commercial scale-up of complex pharmaceutical intermediates, ensuring a steady supply of high-quality materials for downstream drug synthesis.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditional routes to similar chiral hydroxy-methoxy scaffolds often rely on aggressive alkylation conditions or multi-step protection-deprotection sequences that erode overall yield. Conventional methylation strategies frequently employ strong bases like sodium hydride or potassium tert-butoxide, which pose significant safety risks on a large scale and can lead to epimerization of sensitive chiral centers. Furthermore, older processes might utilize stoichiometric amounts of toxic heavy metals or require cryogenic conditions that are energy-intensive and difficult to maintain in large reactor vessels. These limitations result in higher production costs, increased waste generation, and potential supply chain disruptions due to the scarcity of specialized reagents. The cumulative effect of these inefficiencies is a final product with a broader impurity profile, necessitating costly and time-consuming purification steps that delay project timelines and increase the cost of goods sold for the final active pharmaceutical ingredient.
The Novel Approach
The methodology disclosed in the patent circumvents these issues through a carefully orchestrated sequence that prioritizes chemoselectivity and operational simplicity. By initiating the synthesis with a mixed anhydride reduction, the process achieves high conversion to the primary alcohol without affecting the existing ester or carbamate protecting groups. The subsequent methylation step utilizes silver oxide, a heterogeneous reagent that facilitates clean ether formation under reflux conditions, eliminating the need for dangerous pyrophoric bases. This approach not only enhances safety but also simplifies workup procedures, as the silver salts can be easily filtered off. The integration of catalytic hydrogenation for Cbz removal followed by immediate Boc protection demonstrates a telescoping strategy that minimizes isolation steps and solvent usage. This novel approach ensures that the chiral integrity of the molecule is preserved throughout the synthesis, delivering a product suitable for stringent pharmaceutical applications.
Mechanistic Insights into Chemo-selective Reduction and Etherification
The core of this synthetic strategy lies in the precise control of reactivity at each functional group. In the first step, the activation of the carboxylic acid with isobutyl chloroformate and N-methylmorpholine generates a mixed anhydride intermediate. This species is highly electrophilic yet sufficiently stable at low temperatures (-10°C to 0°C) to allow for controlled nucleophilic attack by the hydride source. The subsequent addition of sodium borohydride in methanol at -40°C to -20°C ensures that the reduction proceeds selectively to the alcohol, avoiding over-reduction or transesterification side reactions. This temperature gradient is critical for suppressing the formation of dialkylated byproducts and maintaining the optical purity of the (R)-center. The mechanistic pathway avoids the formation of reactive acyl halides that could lead to racemization, a common pitfall in amino acid derivative synthesis.
Following the reduction, the introduction of the methoxy group via silver oxide-mediated alkylation is a standout feature of this process. Silver oxide acts as a mild base to deprotonate the newly formed hydroxyl group, generating an alkoxide that is immediately trapped by methyl iodide. Unlike homogeneous bases, the heterogeneous nature of silver oxide prevents localized high pH environments that could trigger elimination reactions or ester hydrolysis. The reaction is conducted in acetonitrile at elevated temperatures (70-90°C), which drives the equilibrium towards the ether product while keeping the reaction mixture manageable. Subsequent steps involving acid-catalyzed ester exchange and palladium-catalyzed hydrogenation further refine the molecular architecture. The hydrogenolysis of the benzyloxycarbonyl (Cbz) group is particularly efficient, occurring cleanly at moderate temperatures (45-55°C) to reveal the free amine, which is then instantly protected as the Boc derivative to prevent polymerization or oxidation. This sequence exemplifies a deep understanding of orthogonal protecting group chemistry tailored for industrial feasibility.
How to Synthesize (R)-4-hydroxy-1-methoxybutyl Carbamate Efficiently
Implementing this synthesis requires strict adherence to the temperature profiles and reagent addition rates specified in the patent to ensure reproducibility and safety. The process begins with the activation of the starting acid, followed by a controlled reduction that sets the stage for the entire sequence. Operators must monitor the reaction progress via TLC or HPLC to confirm complete consumption of intermediates before proceeding to the next step, particularly during the methylation and hydrogenation phases where incomplete conversion can lead to difficult-to-remove impurities. The final reduction of the ethyl ester to the primary alcohol using sodium borohydride in a THF/methanol system is exothermic and requires careful thermal management to prevent runaway reactions. Detailed standard operating procedures regarding quenching, extraction, and crystallization are essential to isolate the final product with the required purity specifications.
- Reduce the starting carboxylic acid to the corresponding alcohol using a mixed anhydride method followed by sodium borohydride reduction at low temperatures.
- Perform methylation of the hydroxyl group using silver oxide and methyl iodide in acetonitrile under reflux conditions.
- Execute ester exchange with ethanol/HCl, followed by catalytic hydrogenation to remove the Cbz protecting group and subsequent Boc protection.
- Conclude with the reduction of the ethyl ester to the primary alcohol using sodium borohydride in methanol/THF to yield the final product.
Commercial Advantages for Procurement and Supply Chain Teams
From a procurement and supply chain perspective, this synthetic route offers substantial advantages by utilizing commodity chemicals and avoiding proprietary catalysts. The reliance on reagents such as sodium borohydride, methyl iodide, and di-tert-butyl dicarbonate ensures that raw material sourcing is stable and cost-effective, reducing the risk of supply bottlenecks. The elimination of cryogenic requirements below -40°C for extended periods significantly lowers energy consumption and capital expenditure on specialized cooling equipment, translating to direct cost reduction in pharmaceutical intermediate manufacturing. Furthermore, the high yields reported in the intermediate steps, particularly the quantitative conversions observed in the ester exchange and deprotection stages, minimize material loss and waste disposal costs. This efficiency allows for a more competitive pricing structure for the final intermediate, making it an attractive option for cost-sensitive generic drug programs.
- Cost Reduction in Manufacturing: The process eliminates the need for expensive transition metal catalysts often used in asymmetric synthesis, relying instead on stoichiometric reagents that are inexpensive and readily available globally. By avoiding complex chromatographic purifications in favor of crystallization and extraction workups, the operational expenditure is significantly lowered. The high atom economy of the methylation and protection steps ensures that the majority of the input mass is incorporated into the final product, reducing the volume of chemical waste that requires treatment. These factors combine to create a lean manufacturing process that maximizes output per batch while minimizing overhead costs associated with hazardous material handling and disposal.
- Enhanced Supply Chain Reliability: The use of robust, well-established chemical transformations means that the process can be easily transferred between different manufacturing sites without significant re-validation efforts. The starting materials are bulk commodities with multiple global suppliers, mitigating the risk of single-source dependency. Additionally, the stability of the intermediates allows for potential storage between steps if necessary, providing flexibility in production scheduling. This resilience is crucial for maintaining continuous supply to downstream customers, especially in times of global logistical disruption, ensuring that drug development timelines are not compromised by raw material shortages.
- Scalability and Environmental Compliance: The reaction conditions are inherently scalable, with heat transfer and mixing parameters that are easily managed in standard stainless steel reactors. The avoidance of highly toxic solvents like DMF or HMPA in favor of greener alternatives such as ethanol, ethyl acetate, and acetonitrile aligns with modern environmental, health, and safety (EHS) standards. The simplified workup procedures reduce the load on wastewater treatment facilities, and the ability to recycle solvents like THF and acetonitrile further enhances the sustainability profile of the process. This compliance with green chemistry principles facilitates faster regulatory approval and reduces the environmental footprint of the manufacturing operation.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of this chiral intermediate. Understanding these details is vital for integrating this material into your supply chain and ensuring it meets your specific formulation requirements. The answers are derived directly from the technical disclosures and experimental data provided in the patent literature, offering a transparent view of the process capabilities.
Q: What are the critical temperature controls in the initial reduction step?
A: The initial reduction requires precise temperature control, maintaining the reaction between -10°C and 0°C during mixed anhydride formation, and subsequently lowering to -40°C to -20°C during the sodium borohydride addition to ensure chemoselectivity and prevent side reactions.
Q: How is the stereochemical integrity maintained throughout the six-step sequence?
A: The synthetic route utilizes mild reaction conditions that do not involve strong bases or acids capable of racemizing the chiral center at the alpha-position, ensuring the (R)-configuration is preserved from the starting material to the final carbamate product.
Q: Why is silver oxide used for the methylation step instead of traditional bases?
A: Silver oxide acts as a mild base and promoter for the nucleophilic substitution with methyl iodide, avoiding the harsh conditions associated with strong alkali bases which could compromise the sensitive ester and carbamate functionalities present in the molecule.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable (R)-4-hydroxy-1-methoxybutyl Carbamate Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that high-quality chiral intermediates play in the development of next-generation therapeutics. Our team of expert process chemists has extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from laboratory bench to pilot plant is seamless. We are committed to delivering products with stringent purity specifications, supported by our rigorous QC labs equipped with state-of-the-art analytical instrumentation. Whether you require custom synthesis or large-scale supply of this specific carbamate derivative, our infrastructure is designed to meet the demanding timelines of the global pharmaceutical industry while maintaining the highest standards of quality and safety.
We invite you to contact our technical procurement team to discuss how we can support your project needs. By partnering with us, you gain access to a Customized Cost-Saving Analysis that evaluates the most efficient route for your specific volume requirements. We encourage potential clients to request specific COA data and route feasibility assessments to verify that our manufacturing capabilities align perfectly with your technical expectations. Let us be your strategic partner in bringing innovative medicines to market faster and more efficiently.