Revolutionizing Nucleoside Intermediate Production with Metal-Free Asymmetric Halocyclization
Introduction to Advanced Nucleoside Synthesis Technology
The pharmaceutical industry continuously seeks robust methodologies for constructing complex heterocyclic scaffolds, particularly for antiviral therapeutics. Patent CN113880901B introduces a groundbreaking synthetic approach for preparing (1β,2α,4β) halogenated nucleoside compounds, which serve as pivotal intermediates for furan-ring nucleoside drugs. This technology leverages an asymmetric olefin halocyclization reaction catalyzed by chiral phosphoric acids, marking a significant departure from traditional metal-dependent pathways. By utilizing this organocatalytic strategy, manufacturers can achieve exceptional stereocontrol without the burden of heavy metal contamination. The method demonstrates broad substrate scope and operational simplicity, addressing long-standing challenges in the efficient preparation of β-nucleoside analogs used in treating HIV, HBV, and other viral infections.
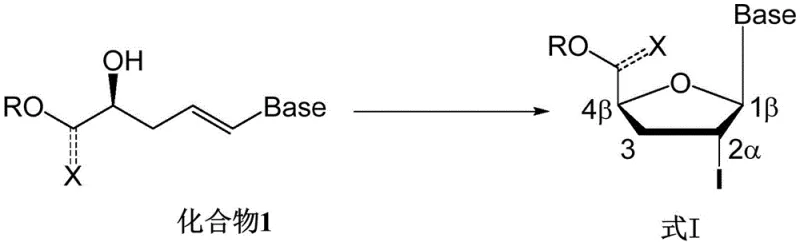
This innovation represents a critical advancement for reliable pharmaceutical intermediates supplier networks aiming to enhance purity profiles. The core transformation involves the cyclization of an alkene-tethered nucleobase precursor into a rigid furanose-like structure with precise stereochemical definition at the C1, C2, and C4 positions. Such precision is paramount for downstream biological activity, as the wrong stereoisomer can lead to reduced efficacy or increased toxicity. The patent details a comprehensive protocol that balances reaction kinetics with thermodynamic stability, ensuring that the desired β-anomer is formed predominantly. This level of control is essential for meeting the stringent regulatory requirements of global health authorities regarding impurity thresholds in active pharmaceutical ingredients.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the construction of glycosidic bonds in nucleoside synthesis has relied heavily on the Vorbrüggen glycosylation reaction. While widely used, this conventional method presents significant drawbacks that hinder efficient manufacturing. The process typically requires stoichiometric amounts of Lewis acids, such as trimethylsilyl trifluoromethanesulfonate (TMSOTf), which creates harsh reaction conditions incompatible with sensitive functional groups. Furthermore, the stereoselectivity of Vorbrüggen glycosylation is notoriously unpredictable, often yielding nearly equimolar mixtures of α and β anomers when C2 substituents are absent or non-participating. For instance, prior art cases in the synthesis of zalcitabine and sofosbuvir have reported α:β selectivity ratios as poor as 40:60 or even 50:50, necessitating cumbersome chromatographic separations that drastically reduce overall yield.
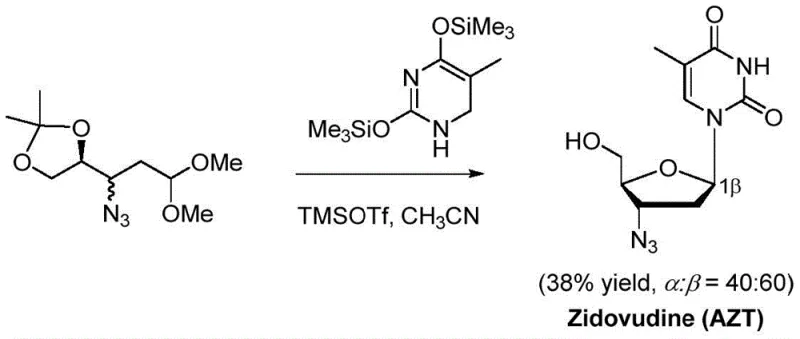
Additionally, the reliance on acyl protecting groups in traditional routes adds unnecessary synthetic steps, including installation and subsequent removal, which lowers atom economy and increases waste generation. The post-processing of these reactions is also difficult on a large scale due to the formation of emulsions and the presence of silicon-containing byproducts. These factors collectively contribute to higher production costs and longer lead times, creating bottlenecks in the supply chain for high-purity pharmaceutical intermediates. The environmental footprint of using stoichiometric Lewis acids and multiple protection-deprotection sequences further complicates compliance with modern green chemistry standards, pushing the industry to seek more sustainable alternatives.
The Novel Approach
In stark contrast, the method disclosed in CN113880901B utilizes an asymmetric halocyclization strategy that circumvents the need for harsh Lewis acids and complex protecting group manipulations. By employing a chiral phosphoric acid catalyst, the reaction proceeds through a highly organized transition state that favors the formation of the desired (1β,2α,4β) configuration. This organocatalytic approach operates under mild conditions, typically between -30°C and 20°C, which preserves the integrity of sensitive nucleobases and side chains. The elimination of metal catalysts not only simplifies the workup procedure but also ensures that the final product is free from toxic metal residues, a critical quality attribute for API manufacturing. The process achieves high diastereomeric ratios (dr values up to 15:1) and excellent isolated yields, often exceeding 85%, thereby maximizing material throughput.
Moreover, this novel route introduces a derivatizable α-halogen substituent at the C2 position, providing a versatile handle for further chemical modifications. This feature allows for the rapid diversification of the nucleoside scaffold to access a wide array of therapeutic candidates through simple functional group transformations. The versatility of the method is evidenced by its compatibility with various nucleobases, including thymine, cytosine, and purine derivatives, as well as different protecting groups on the sugar moiety. This flexibility makes it an ideal platform technology for the commercial scale-up of complex pharmaceutical intermediates, enabling manufacturers to respond quickly to market demands for new antiviral agents while maintaining rigorous quality standards.
Mechanistic Insights into Chiral Phosphoric Acid Catalyzed Halocyclization
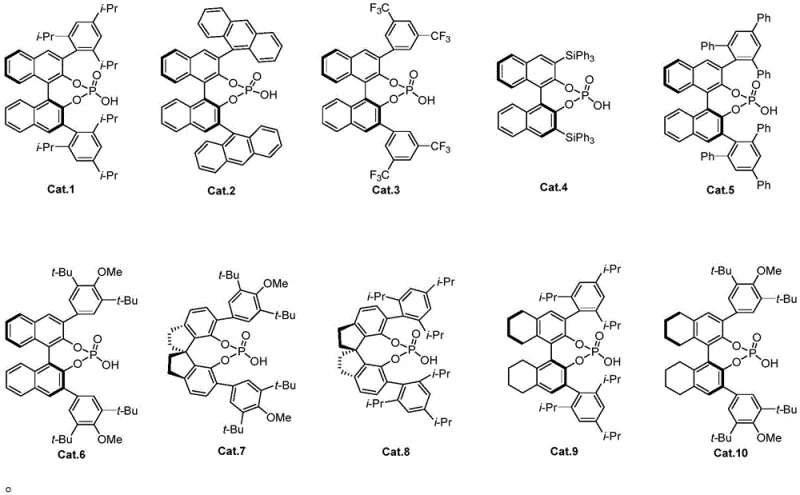
The success of this synthesis hinges on the unique ability of chiral phosphoric acids to activate both the electrophilic halogen source and the nucleophilic olefin substrate simultaneously through hydrogen bonding interactions. The catalyst, typically a binaphthyl-derived phosphoric acid like Cat.9, creates a chiral environment that directs the approach of the halogen species to the alkene double bond. This dual activation mechanism lowers the energy barrier for the cyclization event while imposing strict stereochemical constraints. As the halogen ion attacks the olefin, a cyclic halonium ion intermediate is formed, which is subsequently intercepted by the proximal hydroxyl group or the nucleobase nitrogen, depending on the specific substrate design. The chiral pocket of the catalyst ensures that this ring-closing step occurs exclusively from one face of the molecule, resulting in the high diastereoselectivity observed in the experimental data.
Impurity control is inherently built into this mechanistic pathway due to the high specificity of the organocatalyst. Unlike Lewis acid-catalyzed reactions that can promote non-selective background reactions or rearrangements, the chiral phosphoric acid maintains tight control over the reactive intermediates. This minimizes the formation of regioisomers and stereoisomers that are difficult to separate later in the process. The use of additives such as sodium carbonate or molecular sieves further enhances the reaction efficiency by scavenging acidic byproducts or water that could otherwise deactivate the catalyst or hydrolyze sensitive intermediates. The result is a clean reaction profile that simplifies downstream purification, often requiring only standard column chromatography to achieve pharmaceutical-grade purity. This mechanistic elegance translates directly into operational robustness, making the process highly reproducible across different batches and scales.
How to Synthesize (1β,2α,4β) Halogenated Nucleoside Efficiently
The practical implementation of this synthesis involves a straightforward protocol that can be easily adapted for pilot and commercial production. The detailed standardized synthesis steps are outlined in the guide below, ensuring consistency and safety during operation. Operators must strictly adhere to the specified molar ratios and temperature controls to maintain the high stereoselectivity characteristic of this method. The use of ultra-dry solvents is critical to prevent catalyst deactivation, and the inert atmosphere protects the reactive halogen species from moisture. By following these optimized parameters, manufacturers can reliably produce key intermediates for drugs like stavudine and zalcitabine with superior efficiency compared to legacy methods.
- Dissolve the alkene substrate (Compound 1) and a chiral phosphoric acid catalyst (e.g., Cat.9) in an ultra-dry solvent like chloroform under an inert argon atmosphere.
- Cool the mixture to a temperature range of -78°C to 30°C, preferably around -10°C, and stir for approximately 10 minutes to ensure homogeneity.
- Add additives such as sodium carbonate and a halogen source like N-iodosuccinimide (NIS), then stir for 1 to 72 hours to complete the asymmetric halocyclization.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this organocatalytic technology offers substantial strategic benefits beyond mere technical superiority. The elimination of expensive and toxic transition metal catalysts removes the need for costly metal scavenging steps and extensive analytical testing for residual metals, leading to significant cost reduction in pharmaceutical intermediates manufacturing. The simplified workflow, which avoids multiple protection and deprotection cycles, reduces the consumption of raw materials and solvents, thereby lowering the overall cost of goods sold. Furthermore, the high yields and selectivity minimize waste generation, aligning with corporate sustainability goals and reducing disposal costs associated with hazardous chemical byproducts.
- Cost Reduction in Manufacturing: The metal-free nature of this process eliminates the capital expenditure associated with specialized equipment for metal removal and the recurring cost of scavenger resins. Additionally, the high atom economy and reduced number of synthetic steps mean that less starting material is required to produce the same amount of final product. This efficiency gain directly improves profit margins and allows for more competitive pricing in the global market. The mild reaction conditions also reduce energy consumption for heating or cooling, contributing to lower utility costs over the lifecycle of the product.
- Enhanced Supply Chain Reliability: The reagents used in this method, such as chiral phosphoric acids and N-halosuccinimides, are commercially available from multiple sources, reducing the risk of single-supplier dependency. The robustness of the reaction against minor variations in conditions ensures consistent output, preventing delays caused by batch failures or off-spec material. This reliability is crucial for maintaining continuous supply lines for critical antiviral medications, especially during public health emergencies where demand can surge unexpectedly. The ability to scale this process from grams to tons without losing selectivity provides a secure foundation for long-term supply agreements.
- Scalability and Environmental Compliance: The absence of heavy metals and harsh Lewis acids simplifies the regulatory approval process for new drug applications, as the impurity profile is cleaner and easier to characterize. This facilitates faster time-to-market for new therapies developed using these intermediates. From an environmental perspective, the reduced use of hazardous chemicals and the generation of less toxic waste streamline compliance with increasingly strict environmental regulations. The process is inherently safer for plant operators, reducing the risk of accidents related to exothermic reactions or corrosive reagents, which enhances the overall safety culture of the manufacturing facility.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding this synthesis method, based on the detailed specifications provided in the patent documentation. Understanding these aspects helps stakeholders evaluate the feasibility of integrating this technology into their existing production pipelines. The answers reflect the proven capabilities of the method in delivering high-quality intermediates suitable for GMP manufacturing environments.
Q: What are the advantages of this method over traditional Vorbrüggen glycosylation?
A: Unlike Vorbrüggen glycosylation which often suffers from poor α/β selectivity (e.g., 1:1 ratios) and requires harsh Lewis acids, this organocatalytic method achieves high stereoselectivity (dr values up to 15:1) under mild, metal-free conditions.
Q: Which catalysts are suitable for this asymmetric halocyclization?
A: The process utilizes chiral phosphoric acid small-molecule catalysts. Specifically, binaphthyl-derived phosphoric acids (such as Cat.9 shown in the patent) have demonstrated excellent performance in controlling stereochemistry.
Q: Is this process scalable for commercial API intermediate production?
A: Yes, the method uses commercially available reagents, operates at mild temperatures (-10°C to 20°C), and avoids toxic heavy metals, making it highly suitable for safe and compliant commercial scale-up.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Halogenated Nucleoside Supplier
NINGBO INNO PHARMCHEM stands at the forefront of adopting cutting-edge synthetic technologies to deliver superior value to our global partners. We possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the benefits of this advanced organocatalytic method are fully realized at an industrial level. Our commitment to quality is underscored by our stringent purity specifications and rigorous QC labs, which guarantee that every batch of halogenated nucleoside intermediate meets the highest international standards. By leveraging our expertise in process optimization, we can help you navigate the complexities of nucleoside synthesis with confidence and efficiency.
We invite you to contact our technical procurement team to discuss how this innovative route can optimize your supply chain. Request a Customized Cost-Saving Analysis today to understand the specific economic advantages for your project. Our experts are ready to provide specific COA data and route feasibility assessments tailored to your unique requirements, ensuring a seamless transition to this next-generation manufacturing platform.