Advanced Stereospecific Synthesis of Chiral N-Acylpiperidines for Commercial Scale-Up
Advanced Stereospecific Synthesis of Chiral N-Acylpiperidines for Commercial Scale-Up
The pharmaceutical and fine chemical industries continuously seek robust methodologies for constructing chiral nitrogen heterocycles, which serve as critical scaffolds in bioactive molecules. Patent CN1789247A introduces a groundbreaking method for preparing chiral alpha-substituted or alpha,alpha'-disubstituted N-acylpyrrolidines and N-acylpiperidines. This technology addresses long-standing challenges in alkaloid synthesis by leveraging chiral Betti base-derived intermediates. Unlike conventional routes that struggle with optical purity or require harsh deprotection conditions, this invention offers a streamlined pathway with exceptional stereospecificity and high yields. For R&D directors and procurement specialists, understanding this patent is key to accessing reliable pharmaceutical intermediate supplier networks that can deliver complex chiral amines efficiently. The process eliminates the need for expensive transition metal catalysts in the deprotection step, representing a significant shift towards more sustainable and cost-effective manufacturing paradigms in the production of high-value fine chemicals.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditional synthetic routes for chiral alpha-alkylated piperidines often rely on methods that are operationally complex and economically inefficient. A notable example cited in the background art is the use of chiral glycinol auxiliaries, known as the "CN(R,S)" method. While effective for certain substrates, this approach suffers from significant drawbacks, particularly in the removal of the auxiliary group. The N-debenzylation step typically necessitates metal-catalyzed hydrogenolysis, which requires high-pressure equipment and precious metal catalysts like palladium or platinum. Furthermore, these conditions can sometimes lead to the reduction of other sensitive functional groups within the molecule, limiting substrate scope. Additionally, alternative methods often struggle to maintain high optical purity throughout the synthesis, leading to difficult and costly purification processes to remove enantiomeric impurities. These limitations create bottlenecks in cost reduction in chiral amine manufacturing, making large-scale production less viable for many potential drug candidates.
The Novel Approach
The methodology disclosed in CN1789247A circumvents these issues by utilizing a unique cascade reaction sequence initiated by organometallic addition to chiral oxazine intermediates. The core innovation lies in the use of Betti base derivatives, which possess a diarylbenzylamine structure. This specific structural motif allows for the cleavage of the benzyl C-N bond under mild, non-reducing acidic conditions, rather than requiring catalytic hydrogenation. By reacting the chiral intermediate with Grignard reagents or organolithium compounds, the desired alkyl or alkenyl groups are introduced with high stereocontrol. Subsequent treatment with carboxylic acids or anhydrides facilitates simultaneous N-debenzylation and N-acylation. This dual-functionality step not only simplifies the workflow by combining two transformations but also ensures that the chiral centers remain untouched, preserving the stereochemical integrity of the final product. This approach significantly enhances the commercial scale-up of complex pharmaceutical intermediates by reducing unit operations and safety risks associated with high-pressure hydrogenation.
Mechanistic Insights into Organometallic Alkylation and Acid-Catalyzed Acylation
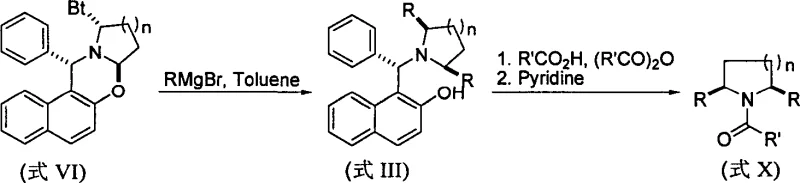
The mechanistic pathway begins with the nucleophilic attack of an organometallic reagent, such as RMgX or RLi, on the electrophilic carbon of the chiral oxazine ring system (Formula I, V, VI, or VII). This addition occurs with high diastereoselectivity due to the rigid conformation imposed by the fused ring system and the steric environment created by the chiral Betti base backbone. The reaction temperature is carefully controlled, typically ranging from 0°C to the boiling point of the solvent (ether, toluene, or THF), to optimize the kinetic control of the addition while preventing side reactions. The resulting intermediate (Formula II, III, or IV) contains the newly formed carbon-carbon bond at the alpha-position of the future piperidine or pyrrolidine ring. This step is crucial for establishing the substitution pattern, whether it be mono-substituted, symmetrically disubstituted, or asymmetrically disubstituted, depending on the specific starting oxazine and the stoichiometry of the organometallic reagent used.
Following the alkylation, the reaction mixture is subjected to a transformative workup involving carboxylic acids and acid anhydrides in the presence of pyridine. The mechanism here relies on the specific electronic properties of the diarylbenzylamine moiety within the intermediate. Under acidic conditions, the benzylic C-N bond becomes labile and undergoes heterolytic cleavage, releasing the benzyl group as a stable carbocation or equivalent species, which is then trapped or decomposed. Simultaneously, the liberated secondary amine reacts with the acid anhydride to form the stable N-acyl derivative. Heating the mixture, preferably at 80°C for approximately 30 minutes, drives this equilibrium to completion and ensures high conversion rates. Since the chiral carbon atoms are not involved in bond-breaking events during this deprotection-acylation cascade, the stereochemistry established in the first step is perfectly retained. This mechanistic elegance ensures high-purity N-acylpiperidine products without the need for chiral resolution steps downstream.
How to Synthesize Chiral N-Acylpiperidines Efficiently
The synthesis protocol outlined in the patent provides a clear roadmap for producing these valuable intermediates with high efficiency. The process is designed to be robust, tolerating a variety of functional groups on the organometallic reagent, including alkenyl chains which are often sensitive to other reaction conditions. The detailed procedure involves precise control of stoichiometry, with organometallic reagents used in excess (1 to 20 equivalents) to ensure complete consumption of the starting oxazine. The subsequent acylation step is equally straightforward, utilizing common reagents like acetic acid, propionic acid, and their corresponding anhydrides. For a comprehensive guide on executing this synthesis in a laboratory or pilot plant setting, please refer to the standardized operating procedures below.
- React chiral Betti base-derived oxazine intermediates (Formula I, V, VI, or VII) with organometallic reagents such as Grignard reagents (RMgX) or organolithium compounds (RLi) in solvents like ether, toluene, or THF at temperatures ranging from 0°C to reflux.
- Subject the resulting alkylated intermediates (Formula II, III, or IV) to a reaction with carboxylic acids or acid anhydrides in the presence of pyridine.
- Heat the mixture, typically around 80°C, to facilitate simultaneous N-debenzylation and N-acylation, yielding the final chiral alpha-substituted or alpha,alpha'-disubstituted N-acylpiperidine or pyrrolidine products with high stereospecificity.
Commercial Advantages for Procurement and Supply Chain Teams
From a procurement and supply chain perspective, the adoption of this synthesis method offers substantial strategic benefits. The elimination of transition metal catalysts for the deprotection step removes a major cost driver and supply chain vulnerability associated with precious metals like palladium. Furthermore, the avoidance of high-pressure hydrogenation equipment reduces capital expenditure requirements for manufacturing facilities and lowers operational safety risks. The use of readily available Grignard reagents and simple carboxylic acids ensures that raw material sourcing is stable and cost-effective. This stability is crucial for maintaining continuous supply lines for high-purity pharmaceutical intermediates, especially in a volatile global market. The simplified workup procedures, which often involve standard aqueous extractions and crystallization or chromatography, further contribute to reducing the overall manufacturing footprint and waste generation.
- Cost Reduction in Manufacturing: The most significant economic advantage stems from the replacement of catalytic hydrogenation with acid-catalyzed cleavage. Transition metal catalysts are not only expensive to purchase but also require rigorous removal steps to meet regulatory limits for residual metals in pharmaceutical products. By avoiding these metals entirely in the deprotection phase, the process drastically simplifies purification, reducing solvent usage and processing time. Additionally, the high yields reported in the examples, often exceeding 80% and reaching up to 93%, mean that less starting material is wasted, directly improving the cost of goods sold (COGS). The ability to perform the reaction in common solvents like ether, toluene, and THF further aligns with standard industrial capabilities, avoiding the need for specialized solvent infrastructure.
- Enhanced Supply Chain Reliability: The reliance on commodity chemicals such as acetic anhydride, pyridine, and standard Grignard reagents ensures a resilient supply chain. Unlike specialized chiral catalysts or enzymes that may have limited suppliers and long lead times, the reagents for this process are produced globally at massive scales. This abundance mitigates the risk of supply disruptions. Moreover, the robustness of the reaction conditions, which tolerate a range of temperatures and do not require inert atmospheres as strictly as some sensitive catalytic cycles, makes the process easier to transfer between different manufacturing sites. This flexibility allows supply chain managers to diversify their production base, ensuring reducing lead time for high-purity intermediates and maintaining consistent delivery schedules to downstream API manufacturers.
- Scalability and Environmental Compliance: Scalability is inherent in the design of this chemistry. The exothermic nature of the Grignard addition is manageable with standard cooling protocols, and the subsequent heating step for acylation is easily controlled in large reactors. From an environmental standpoint, the process generates fewer heavy metal wastes, simplifying effluent treatment and disposal compliance. The high atom economy of the addition step and the efficient conversion in the acylation step minimize the generation of organic byproducts. This aligns well with modern green chemistry principles and increasingly stringent environmental regulations in chemical manufacturing hubs. The ability to produce both alkane and alkene substituted derivatives expands the utility of the platform, allowing a single facility to produce a diverse portfolio of intermediates without significant retooling.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this patented technology. These insights are derived directly from the experimental data and claims presented in CN1789247A, providing clarity on the scope and limitations of the method. Understanding these details is essential for technical teams evaluating the feasibility of integrating this route into their existing production pipelines. The answers highlight the versatility of the method regarding substrate scope and the specific conditions required to achieve optimal results.
Q: What is the primary advantage of this synthesis method over traditional hydrogenolysis?
A: The primary advantage is the ability to perform N-debenzylation under non-reducing, acid-catalyzed conditions. Traditional methods often require expensive transition metal catalysts and high-pressure hydrogenation, which can be hazardous and costly. This patent utilizes the unique structure of Betti base derivatives (diarylbenzylamines) that allow cleavage of the benzyl C-N bond using simple carboxylic acids, significantly simplifying the process and reducing equipment costs.
Q: Does this method maintain stereochemical integrity during the reaction?
A: Yes, the method exhibits high stereospecificity. The reaction proceeds through rigid oxazine intermediates derived from chiral Betti bases. The organometallic addition occurs with high stereocontrol at the alpha-position of the piperidine or pyrrolidine ring. Furthermore, the subsequent N-debenzylation and acylation steps do not involve the chiral carbon atoms, ensuring that the stereochemistry established in the first step is retained intact in the final N-acyl product.
Q: What types of substituents can be introduced using this protocol?
A: The protocol is highly versatile regarding the substituents introduced at the alpha positions. It supports the introduction of a wide range of groups including alkyl (methyl, propyl, nonyl), alkenyl (vinyl, allyl, butenyl), cycloalkyl, and aryl (benzyl) groups. This flexibility allows for the synthesis of diverse libraries of chiral piperidine and pyrrolidine derivatives, making it suitable for generating various alkaloid scaffolds and pharmaceutical intermediates.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Chiral N-Acylpiperidine Supplier
NINGBO INNO PHARMCHEM stands at the forefront of translating advanced academic and patent research into commercial reality. With extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, we possess the technical expertise to implement the sophisticated chemistry described in CN1789247A. Our facilities are equipped to handle the specific requirements of organometallic reactions and acid-catalyzed processes safely and efficiently. We adhere to stringent purity specifications and operate rigorous QC labs to ensure that every batch of chiral N-acylpiperidine or pyrrolidine meets the highest standards required for pharmaceutical applications. Our commitment to quality ensures that the stereochemical integrity and chemical purity of your intermediates are never compromised.
We invite you to collaborate with us to leverage this innovative synthesis method for your next project. Our technical procurement team is ready to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements and target specifications. By partnering with us, you gain access to our deep reservoir of process knowledge and supply chain capabilities. Please contact us today to request specific COA data and route feasibility assessments. Let us help you optimize your supply chain and accelerate your development timeline with our reliable chiral N-acylpiperidine supply solutions.