Mastering 6'-N-Ethyl Gentamicin C1a Synthesis for Advanced Antibiotic Quality Control
The pharmaceutical industry's relentless pursuit of higher safety standards for aminoglycoside antibiotics has brought significant attention to the precise control of impurity profiles in bulk drug substances. Patent CN110498823B introduces a groundbreaking synthetic methodology for producing 6'-N-ethyl gentamicin C1a, a notoriously difficult-to-obtain impurity reference standard essential for the quality assurance of Etimicin Sulfate. This specific compound, previously unavailable as a commercial standard, represents a critical bottleneck in the analytical validation of next-generation semi-synthetic antibiotics. By establishing a reliable chemical pathway to synthesize this molecule, manufacturers can now implement rigorous HPLC monitoring to ensure that residual impurities remain well below toxicological thresholds. The structural complexity of this aminoglycoside derivative requires a sophisticated approach to regioselective functionalization, which this patent successfully addresses through a multi-step protection and modification strategy. 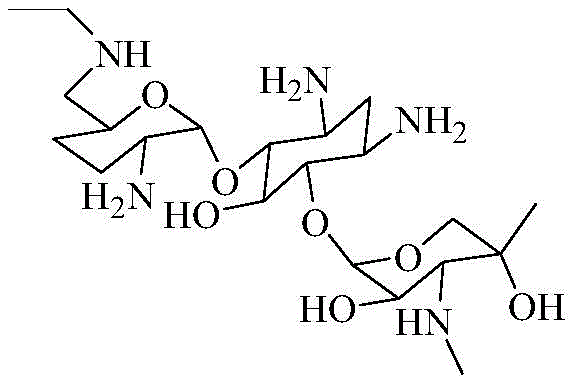 For R&D directors overseeing antibiotic development, access to such high-fidelity reference materials is not merely a regulatory checkbox but a fundamental requirement for demonstrating batch-to-batch consistency and patient safety in global markets.
For R&D directors overseeing antibiotic development, access to such high-fidelity reference materials is not merely a regulatory checkbox but a fundamental requirement for demonstrating batch-to-batch consistency and patient safety in global markets.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditionally, obtaining specific aminoglycoside impurities like 6'-N-ethyl gentamicin C1a relied heavily on isolation from fermentation broths or non-specific degradation of the parent drug, processes fraught with inconsistency and low yield. The natural heterogeneity of fermentation products means that specific minor components often exist in trace amounts, making their isolation via preparative chromatography economically prohibitive and technically exhausting. Furthermore, attempting to synthesize this molecule using non-selective alkylation methods typically results in a complex mixture of N-ethylated isomers at various positions on the sugar rings, necessitating arduous separation procedures that drastically reduce overall throughput. The lack of a dedicated synthetic route has historically forced quality control laboratories to operate without authentic standards, relying instead on relative retention times which compromises the accuracy of impurity quantification. This uncertainty poses a significant risk during regulatory audits, where the inability to definitively identify and quantify known genotoxic or nephrotoxic impurities can lead to costly production delays or even product recalls. Consequently, the industry has urgently needed a deterministic chemical synthesis that bypasses the randomness of biological variation.
The Novel Approach
The methodology disclosed in the patent data revolutionizes this landscape by employing a rational design of orthogonal protecting groups to direct the ethylation specifically to the 6'-amino position. By initially masking the more reactive amines with a tert-butoxycarbonyl (Boc) group under controlled cryogenic conditions, the synthesis effectively shuts down competing reaction pathways that would otherwise lead to unwanted byproducts. This strategic protection allows subsequent acetylation steps to proceed with high regioselectivity, setting the stage for the critical reductive amination step where the ethyl group is introduced. Unlike brute-force alkylation, this route utilizes acetaldehyde and potassium borohydride in a controlled environment to ensure that the ethyl moiety is installed precisely where needed without disturbing the sensitive glycosidic bonds. The result is a streamlined process that transforms a scarce natural product into a readily available synthetic intermediate, decoupling the supply of reference standards from the vagaries of fermentation yields. For procurement managers, this shift from extraction to synthesis signals a move towards more predictable costing and stable supply chains for critical quality control materials.
Mechanistic Insights into Selective Protection and Reductive Amination
The core chemical innovation lies in the meticulous orchestration of protection and deprotection cycles that manipulate the reactivity of the multiple amine functionalities present in the gentamicin C1a scaffold. The process initiates with the reaction of Gentamicin C1a with BOC-ON in a tetrahydrofuran and water system at temperatures strictly maintained between -5°C and 0°C. This low-temperature regime is crucial for kinetic control, favoring the formation of the 6'-N-Boc derivative while minimizing side reactions at the 1 and 3 positions. Following this, the intermediate undergoes acetylation using acetic anhydride and triethylamine in methanol, which converts the remaining free amines into acetamides, effectively locking the molecular architecture into a defined state. The subsequent removal of the Boc group using hydrochloric acid reveals the primary amine at the 6' position exclusively, ready for the final transformation. 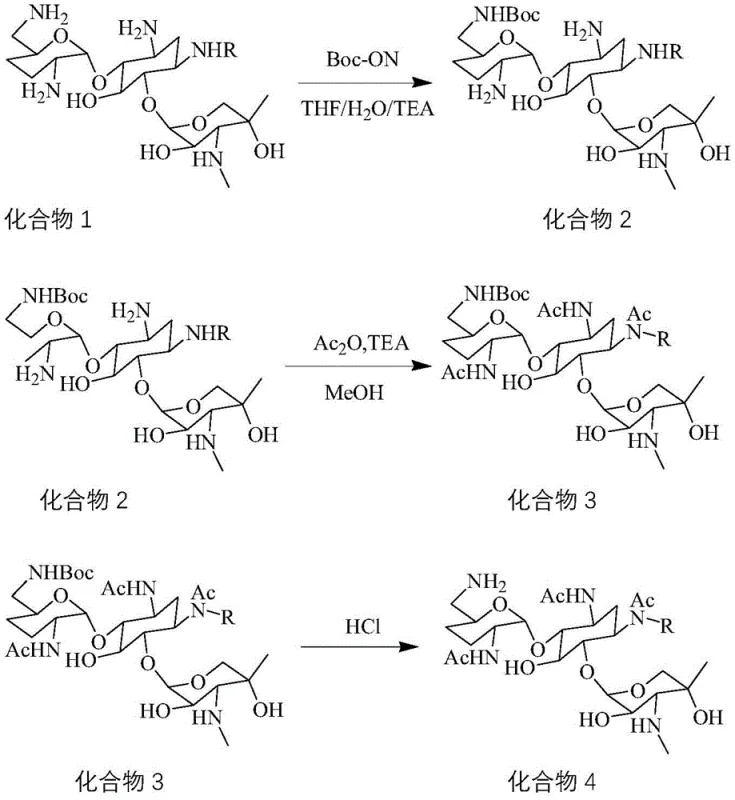 This level of mechanistic precision ensures that the final product is chemically identical to the target impurity found in Etimicin Sulfate batches, providing a true match for analytical comparison. The ability to toggle specific functional groups on and off without affecting the rest of the molecule demonstrates a high degree of synthetic maturity suitable for GMP environments.
This level of mechanistic precision ensures that the final product is chemically identical to the target impurity found in Etimicin Sulfate batches, providing a true match for analytical comparison. The ability to toggle specific functional groups on and off without affecting the rest of the molecule demonstrates a high degree of synthetic maturity suitable for GMP environments.
The final stage of the synthesis involves a reductive amination sequence that installs the ethyl group with remarkable efficiency. Once the 6'-amine is exposed, it reacts with acetaldehyde to form an imine intermediate, which is immediately reduced by potassium borohydride to the secondary amine. This step is conducted in dichloromethane with careful pH buffering to prevent over-alkylation or degradation of the acid-sensitive glycosidic linkages. Following the alkylation, a vigorous hydrolysis step using sodium hydroxide removes the acetyl protecting groups, restoring the native hydroxyl and amine functionalities of the gentamicin core. 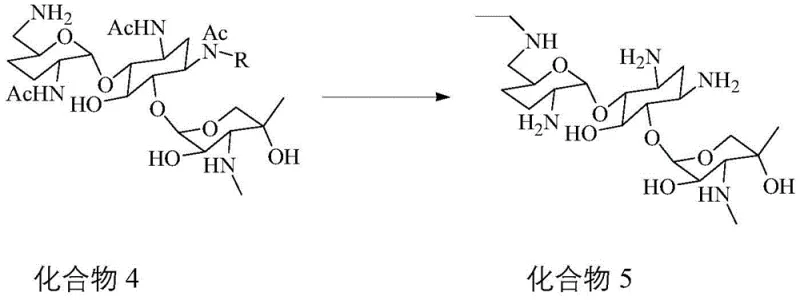 The crude product is then subjected to a sophisticated purification regimen involving macroporous resin D101, which effectively removes inorganic salts and organic byproducts through gradient elution with ethanol. This combination of chemical selectivity and physical purification results in a final product with purity levels reaching up to 99%, as validated by preparative liquid chromatography, setting a new benchmark for impurity standard manufacturing.
The crude product is then subjected to a sophisticated purification regimen involving macroporous resin D101, which effectively removes inorganic salts and organic byproducts through gradient elution with ethanol. This combination of chemical selectivity and physical purification results in a final product with purity levels reaching up to 99%, as validated by preparative liquid chromatography, setting a new benchmark for impurity standard manufacturing.
How to Synthesize 6'-N-Ethyl Gentamicin C1a Efficiently
Implementing this synthesis in a production environment requires strict adherence to the stoichiometric ratios and temperature profiles outlined in the patent to ensure reproducibility and safety. The process begins with the dissolution of the starting material in a ternary solvent system, followed by the controlled addition of the protecting agent to manage exothermicity. Subsequent steps involve precise pH adjustments and solvent swaps, culminating in a high-temperature reflux for deprotection that must be monitored to prevent thermal degradation. The integration of macroporous resin chromatography as a primary purification step offers a scalable alternative to traditional silica gel columns, significantly reducing solvent consumption and processing time. For technical teams looking to adopt this route, the availability of detailed operational parameters provides a clear roadmap from laboratory bench to pilot plant, minimizing the risk of scale-up failures.
- Dissolve Gentamicin C1a in THF/TEA/Water, cool to -5-0°C, and react with BOC-ON to form the 6'-N-Boc protected intermediate.
- Acetylate the protected intermediate using acetic anhydride and triethylamine in methanol to generate the triacetyl derivative.
- Remove the Boc protecting group using hydrochloric acid, adjust pH, and purify via silica gel column chromatography.
- Perform reductive amination with acetaldehyde and potassium borohydride, followed by hydrolysis and macroporous resin purification.
Commercial Advantages for Procurement and Supply Chain Teams
From a commercial perspective, the transition to this synthetic route offers profound advantages in terms of cost stability and supply security for pharmaceutical manufacturers. By eliminating the dependency on fermentation-derived sources, companies can mitigate the risks associated with biological variability, seasonal fluctuations, and contamination events that often plague natural product extraction. The use of commodity chemicals such as acetic anhydride, acetaldehyde, and common solvents ensures that raw material costs remain low and predictable, shielding the supply chain from the volatility of specialized reagent markets. Furthermore, the high specificity of the reaction reduces the burden on downstream purification, leading to significant savings in solvent usage and waste disposal costs. This efficiency translates directly into a more competitive pricing structure for the final impurity standard, allowing quality control budgets to be optimized without compromising on data integrity. For supply chain heads, the robustness of this chemical process guarantees consistent lead times and reliable inventory levels, crucial for maintaining uninterrupted production schedules of the final antibiotic drug.
- Cost Reduction in Manufacturing: The synthetic pathway leverages inexpensive, widely available reagents and avoids the need for expensive transition metal catalysts or proprietary enzymes, resulting in a drastically simplified cost structure. The elimination of complex extraction protocols reduces energy consumption and labor hours, while the high yield of the reductive amination step minimizes raw material waste. These factors combine to lower the overall cost of goods sold, enabling more aggressive pricing strategies in the competitive reference standard market.
- Enhanced Supply Chain Reliability: Unlike fermentation processes that require weeks of growth time and are susceptible to biological failure, this chemical synthesis can be completed in a matter of days with high predictability. The reliance on stable chemical intermediates rather than living organisms ensures that production can be ramped up quickly to meet sudden spikes in demand without the long lead times associated with bioprocessing. This agility provides a strategic buffer against supply disruptions, ensuring that pharmaceutical clients always have access to the critical materials needed for regulatory compliance.
- Scalability and Environmental Compliance: The process is designed with scalability in mind, utilizing unit operations such as liquid-liquid extraction and column chromatography that are easily transferred from laboratory to industrial scale. The use of macroporous resin for desalting reduces the generation of hazardous silica waste, aligning with modern green chemistry principles and simplifying environmental permitting. This eco-friendly profile not only reduces disposal costs but also enhances the corporate sustainability metrics of the manufacturing facility, appealing to environmentally conscious stakeholders.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of this specialized antibiotic intermediate. Understanding the nuances of this synthesis helps stakeholders make informed decisions about integrating this material into their quality control workflows. The answers provided are derived directly from the technical specifications and experimental data contained within the patent documentation, ensuring accuracy and relevance. We encourage potential partners to review these insights to fully appreciate the value proposition of this advanced manufacturing capability.
Q: Why is 6'-N-Ethyl Gentamicin C1a critical for Etimicin Sulfate production?
A: It serves as a critical impurity reference standard required for regulatory quality control, ensuring the safety and efficacy of the final antibiotic formulation by accurately quantifying residual impurities.
Q: What is the achieved purity level of this synthetic method?
A: The optimized synthetic protocol described in patent CN110498823B demonstrates the capability to achieve purity levels exceeding 99% through rigorous macroporous resin desalting and preparative liquid chromatography.
Q: Is this process scalable for commercial intermediate supply?
A: Yes, the method utilizes common reagents like acetic anhydride and potassium borohydride and avoids exotic catalysts, making it highly suitable for scale-up from laboratory grams to multi-kilogram commercial production.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 6'-N-Ethyl Gentamicin C1a Supplier
At NINGBO INNO PHARMCHEM, we understand that the integrity of your antibiotic portfolio depends on the quality of every single component, including the impurity standards used to validate them. Our team of expert chemists has mastered the complex synthesis of 6'-N-Ethyl Gentamicin C1a, leveraging the innovative protocols described in recent patents to deliver materials of unparalleled purity and consistency. We possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that we can meet your demands whether you are in the early research phase or full-scale manufacturing. Our facilities are equipped with rigorous QC labs and stringent purity specifications that exceed industry norms, guaranteeing that every batch we ship is accompanied by comprehensive analytical data. By choosing us as your partner, you gain access to a supply chain that is both resilient and responsive, capable of adapting to the evolving regulatory landscape of the global pharmaceutical industry.
We invite you to collaborate with us to optimize your supply chain for aminoglycoside production and quality control. Our technical procurement team is ready to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements and delivery schedules. We encourage you to reach out today to request specific COA data and route feasibility assessments that demonstrate how our manufacturing excellence can support your commitment to patient safety and product quality. Let us handle the complexities of synthesis so you can focus on delivering life-saving medications to the market with confidence.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →