Optimizing Clevidipine Butyrate Intermediate Production via Novel Dihydropyridine Synthesis
The pharmaceutical industry continuously seeks robust synthetic routes for critical cardiovascular medications, and the preparation of Clevidipine Butyrate intermediates stands as a prime example of this demand. Patent CN103242221A introduces a groundbreaking preparation method for dihydropyridine compounds, specifically targeting the key intermediate required for this potent antihypertensive agent. This technology addresses long-standing challenges in the synthesis of 4-(2',3'-dichlorophenyl)-1,4-dihydro-2,6-dimethyl-3,5-pyridinedicarboxylic acid derivatives, offering a pathway that is not only chemically efficient but also environmentally superior. By leveraging a novel cyclization and deprotection strategy, this method ensures stable product quality and high yields, positioning it as a vital asset for any organization aiming to secure a reliable pharmaceutical intermediates supplier for cardiovascular drug production lines.
Clevidipine Butyrate is a third-generation dihydropyridine calcium channel blocker known for its rapid onset and short duration of action, making it indispensable for acute blood pressure control in hospital settings. The synthesis of its precursors requires precise chemical engineering to maintain the integrity of the dihydropyridine ring while managing sensitive functional groups. The innovation disclosed in this patent provides a streamlined approach that bypasses the hazardous byproducts associated with earlier generations of synthesis technology. For R&D directors and procurement specialists, understanding this shift is crucial, as it directly impacts the cost reduction in API manufacturing and the overall safety profile of the supply chain. The ability to produce high-purity dihydropyridine compounds without generating toxic volatiles represents a significant leap forward in green chemistry and process safety.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of key dihydropyridine intermediates has relied heavily on protecting groups that pose significant safety and environmental hazards during the deprotection phase. Prior art, such as the methods described in patent WO2000/31035 and CN101759631A, frequently utilized 2-cyanoethyl groups to protect carboxylic acid functionalities. While these methods achieved the desired chemical transformation, the removal of the 2-cyanoethyl group invariably generated acrylonitrile as a byproduct. Acrylonitrile is a highly toxic, volatile, and flammable substance that presents severe risks to operator health and requires complex, costly waste treatment infrastructure to manage safely. Furthermore, these conventional routes often suffered from lower overall yields and required multiple recrystallization steps to achieve the necessary purity levels for pharmaceutical applications, thereby increasing production time and material costs significantly.
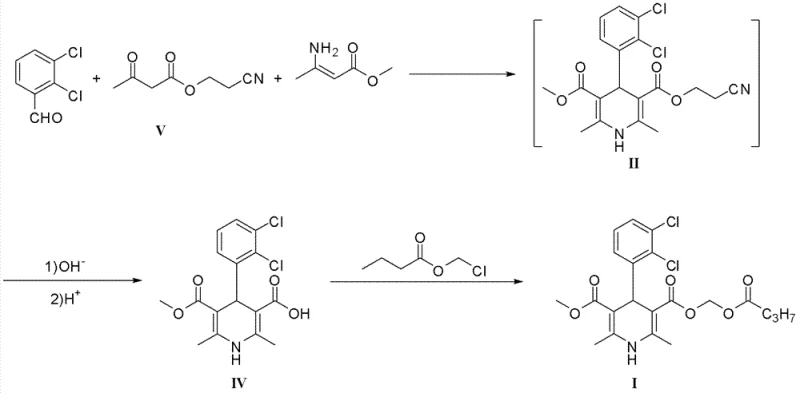
The Novel Approach
In stark contrast to these legacy methods, the novel approach detailed in CN103242221A employs commonly available protecting groups such as tert-butyl, benzyl, or substituted benzyl esters that do not generate toxic substances upon removal. This strategic shift in chemical design eliminates the formation of acrylonitrile entirely, replacing it with benign byproducts that are easily managed during standard workup procedures. The new method involves a condensation reaction between specific aminocrotonate derivatives and dichlorobenzylidene acetoacetates, followed by a mild deprotection step that can be achieved using either acidic conditions or catalytic hydrogenation. This flexibility allows manufacturers to choose the most cost-effective and scalable conditions for their specific facility capabilities. The result is a process that is not only safer and more environmentally compliant but also operationally simpler, reducing the burden on quality control and waste management teams while enhancing the commercial scale-up of complex pharmaceutical intermediates.
Mechanistic Insights into Hantzsch-like Cyclization and Deprotection
The core of this innovative synthesis lies in the efficient construction of the 1,4-dihydropyridine ring system through a modified Hantzsch condensation reaction. The process begins with the reaction of Compound VII, an aminocrotonate ester with a specific protecting group, and Compound VIII, a 2,3-dichlorobenzylidene acetoacetate derivative. Under controlled thermal conditions in alcoholic solvents, these precursors undergo a cyclocondensation to form the protected intermediate IX. The choice of solvent, such as ethanol, isopropanol, or methanol, plays a critical role in solubilizing the reactants and facilitating the transition state required for ring closure. The reaction temperature is carefully maintained between 60°C and 80°C to optimize kinetics without promoting degradation of the sensitive dihydropyridine core. This step is fundamental to establishing the stereochemistry and substitution pattern required for the biological activity of the final antihypertensive agent.
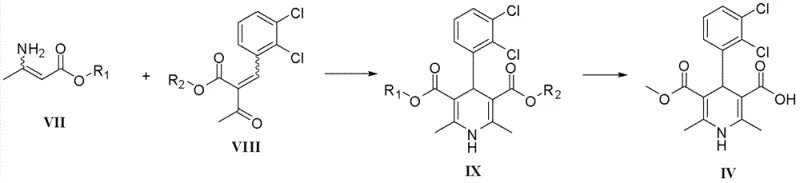
Following the formation of intermediate IX, the deprotection strategy is executed to reveal the free carboxylic acid at the 3-position of the pyridine ring, yielding Compound IV. The patent outlines two distinct mechanistic pathways for this transformation, providing valuable versatility for process chemists. The first pathway involves acidic hydrolysis using reagents like hydrochloric acid or trifluoroacetic acid, which cleaves the ester bond under mild temperatures ranging from -10°C to 50°C. The second pathway utilizes catalytic hydrogenation with palladium on carbon or Raney-Nickel, which is particularly effective for benzyl-type protecting groups. This dual-option mechanism ensures that impurities can be effectively controlled; for instance, hydrogenation can simultaneously reduce unsaturated impurities, leading to a cleaner crude product. The ability to tune the deprotection conditions allows for precise impurity profile management, ensuring that the final high-purity dihydropyridine compounds meet stringent regulatory specifications for clinical use.
How to Synthesize Clevidipine Intermediate Efficiently
Implementing this synthesis route requires careful attention to stoichiometry and reaction monitoring to maximize the yield of the protected intermediate before proceeding to deprotection. The molar ratio of the aminocrotonate to the benzylidene acetoacetate is typically maintained between 1:1 and 1:1.5 to ensure complete consumption of the limiting reagent while minimizing excess waste. Post-reaction processing involves simple concentration and, in some embodiments, crystallization directly from the reaction mixture, which significantly reduces solvent usage and processing time compared to chromatographic separations. The standardized synthesis steps below outline the critical parameters for replicating this high-efficiency process in a pilot or production environment, ensuring consistency and reproducibility across batches.
- Perform a ring closure reaction between compound VII and compound VIII in alcoholic solvents at 60-80°C to form intermediate IX.
- Execute deprotection of intermediate IX using acidic reagents or catalytic hydrogenation to obtain the final carboxylic acid compound IV.
- Isolate the product through filtration or crystallization, ensuring removal of catalyst residues and solvent traces for high purity.
Commercial Advantages for Procurement and Supply Chain Teams
From a commercial perspective, the adoption of this patented methodology offers substantial advantages that extend beyond mere chemical efficiency, directly addressing the pain points of procurement managers and supply chain heads. The elimination of toxic acrylonitrile generation removes the need for specialized scrubbing systems and hazardous waste disposal contracts, leading to significant cost reduction in API manufacturing. Furthermore, the use of inexpensive and readily available reagents such as common alcohols and mineral acids stabilizes the raw material supply chain, reducing vulnerability to market fluctuations. The simplicity of the post-treatment process, often requiring only filtration and washing rather than complex chromatography, drastically shortens the production cycle time. This efficiency translates into enhanced supply chain reliability, allowing manufacturers to respond more quickly to market demand for cardiovascular medications without compromising on quality or safety standards.
- Cost Reduction in Manufacturing: The economic benefits of this process are driven by the replacement of expensive or hazardous reagents with commodity chemicals and the simplification of downstream processing. By avoiding the generation of toxic byproducts, facilities save considerably on environmental compliance costs and waste treatment fees. Additionally, the high yields reported in the patent examples mean that less raw material is required to produce the same amount of active intermediate, improving the overall material cost efficiency. The ability to use catalytic hydrogenation for deprotection also allows for the recovery and reuse of catalysts in some configurations, further driving down the variable costs associated with large-scale production runs.
- Enhanced Supply Chain Reliability: Supply chain continuity is bolstered by the reliance on widely available starting materials that are not subject to the same regulatory restrictions as toxic precursors. The robustness of the reaction conditions, which tolerate a range of temperatures and solvents, reduces the risk of batch failures due to minor process deviations. This resilience ensures a steady flow of high-purity dihydropyridine compounds to downstream formulation partners. Moreover, the reduced complexity of the synthesis minimizes the number of unit operations required, decreasing the potential for equipment bottlenecks and maintenance downtime. This operational stability is critical for maintaining long-term supply agreements with major pharmaceutical companies.
- Scalability and Environmental Compliance: Scaling this process from laboratory to industrial production is facilitated by the absence of hazardous gas evolution and the use of standard reactor types. The environmental footprint of the manufacturing process is significantly reduced, aligning with global sustainability goals and stricter environmental regulations. The waste streams generated are primarily organic solvents and salts, which are easier to treat and recycle compared to cyanide-containing wastes. This environmental compatibility not only mitigates regulatory risk but also enhances the corporate social responsibility profile of the manufacturer. The process is designed to be inherently safe, reducing the need for extensive safety interlocks and allowing for more flexible plant scheduling and capacity utilization.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this dihydropyridine synthesis technology. These answers are derived directly from the experimental data and claims presented in the patent documentation, providing a factual basis for decision-making. Understanding these details helps stakeholders evaluate the feasibility of integrating this route into their existing manufacturing portfolios. The focus remains on practical considerations such as safety, yield, and scalability, which are paramount for successful commercialization.
Q: How does this method improve safety compared to cyanoethyl protection?
A: Unlike prior art using 2-cyanoethyl groups which release toxic acrylonitrile during deprotection, this patent utilizes common protecting groups like tert-butyl or benzyl that degrade into harmless byproducts, significantly enhancing operational safety.
Q: What are the typical yield improvements observed in this synthesis?
A: The patent reports high yields across multiple examples, with specific embodiments achieving over 70% to 96% yield depending on the protecting group and deprotection method used, ensuring efficient material utilization.
Q: Is this process suitable for large-scale commercial manufacturing?
A: Yes, the process uses inexpensive reagents, common solvents like ethanol and methanol, and simple post-treatment steps such as filtration and crystallization, making it highly scalable for industrial production.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Clevidipine Intermediate Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of securing a stable and high-quality supply of key pharmaceutical intermediates for the global healthcare market. Our team of expert chemists has thoroughly analyzed the technological advancements presented in CN103242221A and possesses the extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production. We are committed to delivering stringent purity specifications and maintaining rigorous QC labs to ensure that every batch of dihydropyridine compound meets the highest international standards. Our infrastructure is designed to handle complex synthetic routes safely and efficiently, ensuring that our partners receive materials that facilitate smooth downstream processing and final drug product manufacturing.
We invite you to collaborate with us to leverage this advanced synthesis technology for your supply chain needs. Our technical procurement team is ready to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements and quality targets. Please contact us to request specific COA data and route feasibility assessments that demonstrate how our capabilities align with your project goals. By partnering with NINGBO INNO PHARMCHEM, you gain access to a reliable pharmaceutical intermediates supplier dedicated to innovation, quality, and long-term partnership success in the competitive landscape of cardiovascular drug development.