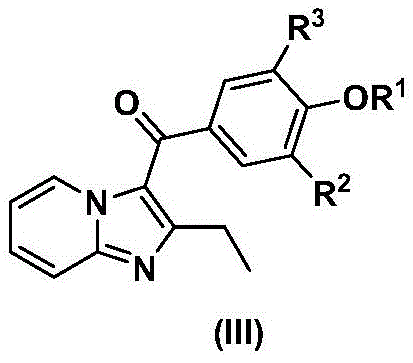
Scalable Synthesis of 3-Bromo-5-(2-ethylimidazo[1,2-a]pyridine-3-carbonyl)-2-hydroxybenzonitrile for Gout Therapy
Introduction to Advanced URAT1 Inhibitor Synthesis
The pharmaceutical landscape for treating metabolic disorders such as gout and hyperuricemia is evolving rapidly, driven by the need for more effective URAT1 inhibitors. Patent CN111410654A discloses a groundbreaking synthetic methodology for 3-bromo-5-(2-ethylimidazo[1,2-a]pyridine-3-carbonyl)-2-hydroxybenzonitrile, a potent candidate capable of inhibiting uric acid reabsorption. This technical insight report analyzes the proprietary process developed to overcome the limitations of prior art, specifically addressing the challenges of low conversion rates and difficult purification associated with traditional iodine-based routes. By shifting to a optimized bromine-based strategy with a unique two-stage cyclization protocol, this technology offers a robust pathway for producing high-purity pharmaceutical intermediates. For R&D directors and procurement specialists, understanding these mechanistic improvements is crucial for securing a reliable pharmaceutical intermediate supplier that can deliver consistent quality at scale.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of complex imidazopyridine derivatives relied heavily on methods described in earlier patents such as CN201610810990, which utilized selective fluorine and iodine substitutions on the benzene ring. These conventional routes suffered from significant drawbacks, primarily due to the reliance on single-solvent heating for the critical ring-closure reaction. This approach often resulted in incomplete reactions and low conversion rates, creating a complex mixture of impurities that were difficult to separate. The necessity for extensive purification, often involving column chromatography, not only increased production costs but also drastically reduced the overall isolation yield. Furthermore, the use of iodine reagents introduced unnecessary expense and handling complexities, making the process less attractive for cost reduction in API manufacturing. These inefficiencies created a bottleneck for clinical supply, highlighting the urgent need for a more streamlined and industrially viable synthetic strategy.
The Novel Approach
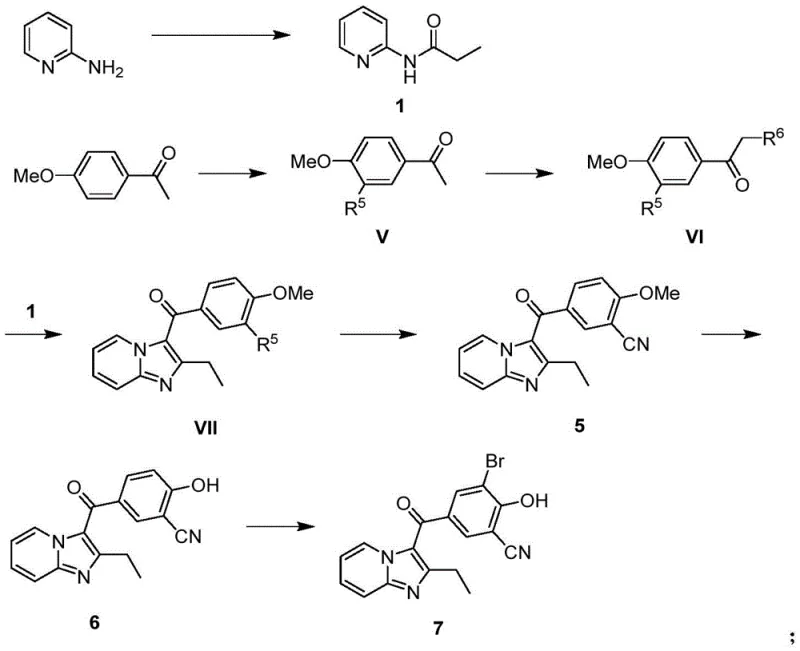
The innovative methodology presented in CN111410654A introduces a paradigm shift by employing a two-stage reaction sequence for the formation of the core imidazopyridine scaffold. Instead of a single heating step, the process initiates with the reaction of the amide and ketone substrates in an organic solvent, followed by a distinct second stage where water and a base are introduced. This strategic addition of base in the second phase, or potentially in both phases, dramatically promotes the conversion of intermediate species into the desired target product while simultaneously suppressing the formation of unwanted by-products. As illustrated in the comprehensive reaction scheme below, this approach allows for the use of more economical bromine reagents instead of iodine, significantly lowering raw material costs. The result is a process that achieves high separation yields through simple recrystallization, eliminating the need for chromatographic purification and enhancing the overall economic feasibility of the synthesis.
Mechanistic Insights into Two-Stage Cyclization and Selective Halogenation
The core innovation of this synthesis lies in the precise control of the cyclization mechanism between the N-(pyridin-2-yl)propanamide and the alpha-halo ketone intermediate. The reaction is divided into two thermal stages: an initial heating phase at 75~150℃ in an organic solvent such as ethyl acetate, followed by a continuation phase at 50~100℃ in the presence of water. The introduction of a base, such as sodium bicarbonate, during the second stage adjusts the pH of the reaction mixture to a range of 4~7. This pH control is critical for facilitating the nucleophilic attack and subsequent ring closure while neutralizing acidic by-products that could otherwise degrade the sensitive imidazopyridine structure. By optimizing the molar ratio of the base to the ketone substrate (preferably 0.8~1.2 equivalents), the process ensures that the equilibrium shifts decisively towards the product, thereby maximizing the yield and minimizing the generation of polymeric or hydrolyzed impurities that typically plague single-stage reactions.
Furthermore, the preparation of the ketone precursor involves a sophisticated two-step halogenation strategy that ensures high regioselectivity. The first halogenation occurs on the benzene ring of 4-methoxyacetophenone under acidic conditions, while the second targets the alpha-position of the acetyl group. This sequential addition of halogenating reagents, such as N-bromosuccinimide (NBS), prevents over-halogenation and ensures that the bromine atoms are placed exactly where needed for the subsequent cyclization and functionalization steps. The ability to control these halogenation events independently allows for the suppression of side reactions that would otherwise lower the purity of the intermediate. This level of mechanistic control is essential for producing high-purity URAT1 inhibitor intermediates that meet the stringent quality standards required for clinical trial materials and eventual commercial drug substance manufacturing.
How to Synthesize 3-Bromo-5-(2-ethylimidazo[1,2-a]pyridine-3-carbonyl)-2-hydroxybenzonitrile Efficiently
The practical execution of this synthesis requires careful attention to solvent selection and temperature gradients to ensure reproducibility and safety. The preferred route utilizes ethyl acetate as the primary organic solvent due to its favorable boiling point and ease of removal, coupled with a workup procedure that relies on aqueous washes and recrystallization rather than complex extraction protocols. The detailed standardized synthetic steps, including specific reagent quantities and reaction times derived from the patent examples, are outlined in the guide below to assist process chemists in replicating this high-yielding pathway.
- Acylation of 2-aminopyridine with propionic anhydride to form N-(pyridin-2-yl)propanamide.
- Selective bromination of 4-methoxyacetophenone followed by alpha-halogenation to generate the key ketone intermediate.
- Two-stage cyclization reaction between the amide and ketone intermediates in ethyl acetate with controlled base addition.
- Cyanation using cuprous cyanide, followed by demethylation and final bromination to yield the target URAT1 inhibitor intermediate.
Commercial Advantages for Procurement and Supply Chain Teams
From a supply chain perspective, this novel synthetic route offers substantial benefits by simplifying the manufacturing workflow and reducing dependency on scarce or expensive reagents. The elimination of column chromatography is a major operational advantage, as it removes a significant bottleneck in production throughput and reduces the consumption of silica gel and solvents. This streamlining directly translates to faster batch turnover times and a more reliable supply of critical intermediates for downstream drug development. Additionally, the use of commercially available starting materials like 4-methoxyacetophenone and 2-aminopyridine ensures that the supply chain remains resilient against raw material shortages, providing procurement managers with greater confidence in long-term sourcing strategies.
- Cost Reduction in Manufacturing: The transition from iodine-based reagents to bromine-based alternatives like NBS represents a significant decrease in raw material expenditure, as bromine sources are generally more abundant and cost-effective. Moreover, the high yield achieved in the cyclization step (reported up to 64.7% in specific examples without optimization) combined with the avoidance of chromatographic purification drastically reduces the cost per kilogram of the final intermediate. The simplified workup procedure also lowers utility costs associated with solvent recovery and waste disposal, contributing to overall cost reduction in API manufacturing.
- Enhanced Supply Chain Reliability: By utilizing robust reaction conditions that tolerate minor variations in temperature and pH, the process demonstrates high resilience, which is critical for maintaining consistent supply. The reliance on standard industrial solvents such as ethyl acetate and NMP, rather than exotic or highly regulated chemicals, further mitigates supply chain risks. This stability ensures that production schedules can be met reliably, reducing lead time for high-purity pharmaceutical intermediates and preventing delays in the clinical development timeline.
- Scalability and Environmental Compliance: The process is inherently designed for commercial scale-up of complex heterocyclic compounds, with reaction temperatures and pressures that are easily manageable in standard stainless steel reactors. The absence of heavy metal catalysts in the key cyclization step simplifies the impurity profile, making it easier to meet regulatory limits for residual metals in the final drug product. Furthermore, the ability to recycle solvents and the reduction in hazardous waste generation align with modern environmental compliance standards, supporting sustainable manufacturing practices.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis route. These answers are derived directly from the experimental data and process descriptions provided in the patent documentation, offering clarity on the feasibility and advantages of the technology for potential partners and licensees.
Q: How does the new two-stage cyclization method improve yield compared to conventional single-solvent heating?
A: The novel two-stage method involves an initial reaction in organic solvent followed by a second stage with water and base. This specifically promotes the conversion of intermediates to the target product while suppressing by-product formation, significantly increasing separation yield and purity without requiring column chromatography.
Q: What are the critical control points for the halogenation steps in this synthesis?
A: The process utilizes two precisely controlled halogenation reactions with different selectivities. The first stage introduces the ring halogen (e.g., bromine) under acidic conditions, while the second stage targets the alpha-position of the ketone. This staged approach prevents over-halogenation and ensures high regioselectivity.
Q: Is this synthesis route suitable for large-scale industrial production?
A: Yes, the route is designed for industrial scalability. It uses readily available raw materials like 4-methoxyacetophenone and avoids expensive catalysts. The purification relies on simple recrystallization and filtration rather than complex chromatography, making it cost-effective and robust for commercial manufacturing.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 3-Bromo-5-(2-ethylimidazo[1,2-a]pyridine-3-carbonyl)-2-hydroxybenzonitrile Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of robust synthetic pathways in the development of next-generation therapeutics for metabolic diseases. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from laboratory discovery to industrial manufacturing is seamless. We are committed to delivering products with stringent purity specifications, supported by our rigorous QC labs that utilize advanced analytical techniques to verify every batch. Our capability to implement the two-stage cyclization technology described in CN111410654A positions us as a strategic partner capable of meeting the demanding quality and volume requirements of global pharmaceutical clients.
We invite you to engage with our technical procurement team to discuss how this optimized synthesis can benefit your specific project needs. By requesting a Customized Cost-Saving Analysis, you can gain deeper insights into the economic advantages of switching to this bromine-based route. We encourage you to contact us today to obtain specific COA data and route feasibility assessments, allowing you to make informed decisions that accelerate your drug development pipeline while optimizing your manufacturing budget.