Scalable Synthesis of Bivalent DNJ Derivatives for Advanced Diabetes Drug Development
Scalable Synthesis of Bivalent DNJ Derivatives for Advanced Diabetes Drug Development
The pharmaceutical industry is constantly seeking novel enzyme inhibitors that offer superior efficacy and pharmacokinetic profiles compared to traditional monomeric drugs. A significant breakthrough in this domain is documented in Chinese Patent CN112441963B, which discloses a robust synthetic route for a novel bivalent state DNJ (1-deoxynojirimycin) derivative. This specific compound, characterized by a terephthaloyl linker connecting two DNJ moieties, represents a strategic evolution in the design of alpha-glucosidase inhibitors. Unlike previous iterations that often suffered from reduced activity when modified for multivalency, this derivative demonstrates a potent IC50 value of 0.302±0.030mM against alpha-glucosidase. The structural integrity and functional positioning of the hydroxyl groups are critical for this bioactivity, as illustrated in the molecular architecture below.
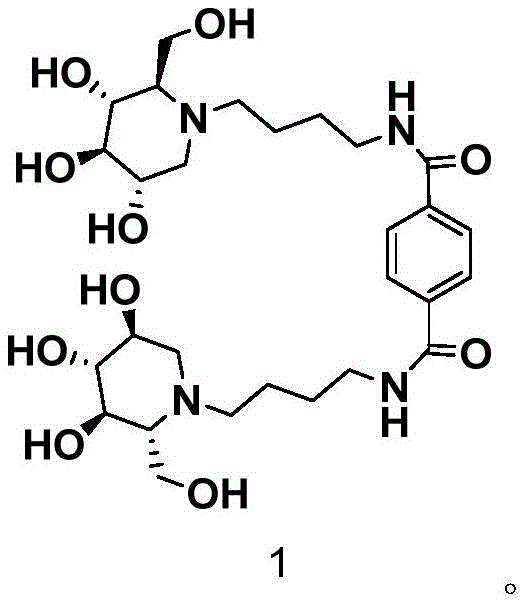
For R&D directors and procurement specialists, understanding the synthesis of such complex imino sugar derivatives is paramount. The patent outlines a concise two-step methodology that avoids the harsh conditions often associated with click chemistry approaches. By leveraging standard amide coupling followed by catalytic hydrogenation, the process ensures high yields and operational simplicity. This technical insight report delves deep into the mechanistic advantages, commercial viability, and supply chain implications of adopting this synthetic pathway for the production of high-purity pharmaceutical intermediates intended for diabetes and obesity therapeutics.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the development of multivalent DNJ derivatives has faced significant hurdles regarding both synthetic complexity and biological efficacy. Many prior art strategies relied heavily on click chemistry reactions to conjugate DNJ active units with various scaffolds. While click chemistry is powerful, it often introduces triazole linkers that can alter the spatial orientation of the pharmacophores, potentially leading to steric hindrance at the enzyme active site. Literature cited in the background of the patent, such as work by Gouin et al., indicates that certain bivalent DNJ derivatives synthesized via these conventional routes exhibited no inhibitory activity against alpha-glucosidase. Furthermore, traditional deprotection methods for imino sugars frequently require strong acids or Lewis acids that can degrade the sensitive piperidine ring or cause epimerization, resulting in difficult-to-remove impurities that compromise the final drug substance quality.
The Novel Approach
The methodology presented in CN112441963B offers a transformative solution by utilizing a rigid terephthaloyl linker introduced via direct acylation. This approach not only simplifies the synthetic sequence but also optimizes the distance between the two DNJ units to match the multivalent binding requirements of the biological target. The reaction proceeds under mild conditions, starting with the coupling of N-(4-aminobutyl)-2,3,4,6-tetrabenzyl-1-deoxynojirimycin and terephthaloyl chloride. The subsequent deprotection is achieved through catalytic hydrogenation rather than harsh chemical cleavage. This entire workflow is depicted in the comprehensive reaction scheme below, highlighting the efficiency of transforming protected precursors into the final bioactive molecule without compromising the stereochemical integrity of the chiral centers.
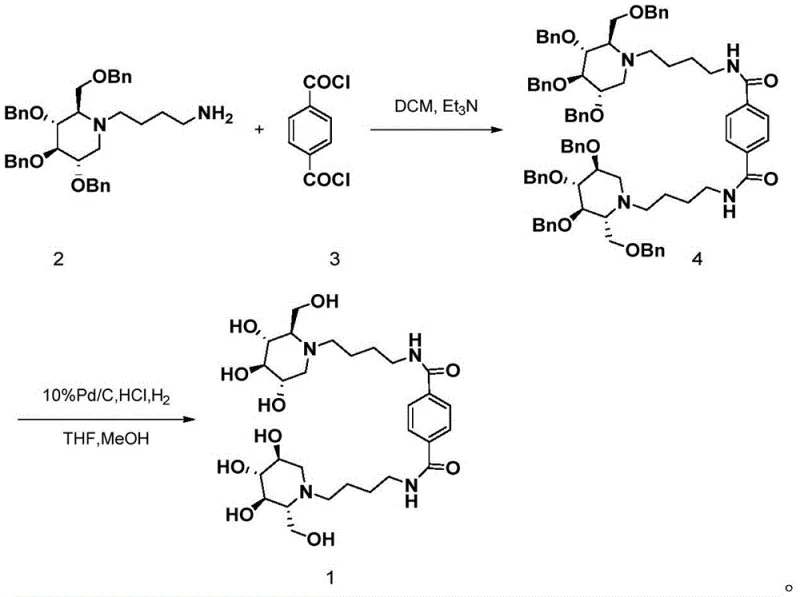
From a process chemistry perspective, this route is exceptionally elegant. It eliminates the need for copper catalysts typically required in click reactions, thereby removing a major source of heavy metal contamination that requires extensive downstream purification. The use of terephthaloyl chloride, a commodity chemical, ensures that the core linker is cost-effective and readily available. The transition from the fully protected intermediate (Compound 4) to the final product (Compound 1) is achieved in a single pot under atmospheric pressure, drastically reducing the energy consumption and equipment complexity associated with high-pressure hydrogenation reactors. This streamlined approach directly addresses the pain points of scalability and cost-efficiency that plague many complex carbohydrate syntheses.
Mechanistic Insights into Amide Coupling and Catalytic Hydrogenation
The first stage of the synthesis involves a nucleophilic acyl substitution reaction where the primary amine of the protected DNJ derivative attacks the carbonyl carbon of the terephthaloyl chloride. This reaction is conducted in dichloromethane with triethylamine serving as an acid scavenger to neutralize the generated HCl. Maintaining the temperature between 0°C and 30°C is crucial to prevent side reactions such as over-acylation or hydrolysis of the acid chloride. The stoichiometry and addition rate are carefully controlled to ensure the formation of the symmetric bis-amide structure. The presence of four benzyl protecting groups on the DNJ ring enhances the solubility of the intermediate in organic solvents, facilitating homogeneous reaction kinetics and simplifying the monitoring of reaction progress via thin-layer chromatography.
The second stage is a classic hydrogenolysis reaction where the benzyl ether protecting groups are cleaved to reveal the free hydroxyl groups essential for enzyme binding. This step utilizes 10% Pd/C as a heterogeneous catalyst under a hydrogen atmosphere of 1-2 atm. A critical mechanistic detail is the addition of hydrochloric acid to adjust the pH of the reaction system to between 2 and 4. This acidic environment serves a dual purpose: it protonates the tertiary amine nitrogen of the DNJ ring, preventing it from coordinating with and poisoning the palladium catalyst, and it facilitates the cleavage of the benzyl-oxygen bond. The solvent system, a mixture of tetrahydrofuran and methanol, provides the necessary polarity to dissolve the polar intermediate while maintaining compatibility with the catalyst. This precise control over reaction parameters ensures high conversion rates and minimizes the formation of partially deprotected byproducts.
How to Synthesize Bivalent DNJ Derivative Efficiently
Executing this synthesis requires strict adherence to the optimized parameters defined in the patent to achieve the reported yields of 80% for the coupling step and 71.9% for the deprotection step. The process begins with the preparation of the protected amine precursor, which must be dry and free of impurities to ensure efficient coupling. The reaction mixture is stirred for approximately 7 hours at room temperature after an initial cooling period, allowing sufficient time for the diffusion-controlled coupling to reach completion. Following the isolation of the intermediate, the hydrogenation step is performed at room temperature for 6 hours. The workup involves simple filtration through celite to remove the palladium catalyst, followed by concentration and purification. For a detailed breakdown of the standardized operating procedures and safety protocols required for this synthesis, please refer to the technical guide below.
- React N-(4-aminobutyl)-2,3,4,6-tetrabenzyl-1-deoxynojirimycin with terephthaloyl chloride in DCM and triethylamine at 0°C to room temperature for 7 hours.
- Perform debenzylation using 10% Pd/C catalyst under 1-2 atm hydrogen pressure in THF/MeOH with pH adjustment to 2-4 using HCl.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this synthetic route offers tangible benefits that extend beyond mere chemical elegance. The reliance on commodity reagents like terephthaloyl chloride and standard solvents like DCM and methanol means that the raw material supply chain is robust and less susceptible to geopolitical disruptions or niche vendor bottlenecks. Furthermore, the elimination of specialized click chemistry reagents and copper catalysts significantly reduces the cost of goods sold (COGS). The simplified purification process, which avoids complex heavy metal scavenging steps, translates into shorter cycle times and higher throughput in manufacturing facilities. These factors collectively contribute to a more resilient and cost-effective supply chain for producing high-purity pharmaceutical intermediates.
- Cost Reduction in Manufacturing: The process eliminates the need for expensive transition metal catalysts often used in alternative coupling strategies, such as copper for click chemistry, which require rigorous and costly removal steps to meet pharmaceutical standards. By utilizing palladium on carbon for hydrogenation, a standard and recoverable catalyst, the overall material costs are significantly lowered. Additionally, the high yields reported in the patent minimize waste generation and maximize the utilization of the valuable DNJ starting material, leading to substantial cost savings in raw material consumption.
- Enhanced Supply Chain Reliability: The starting materials, including terephthaloyl chloride and protected DNJ derivatives, are commercially available from multiple global suppliers, reducing the risk of single-source dependency. The reaction conditions are mild and do not require specialized high-pressure or cryogenic equipment, meaning that production can be easily transferred between different contract manufacturing organizations (CMOs) without significant capital investment. This flexibility ensures continuous supply continuity even in the face of facility maintenance or unexpected downtime at specific manufacturing sites.
- Scalability and Environmental Compliance: Operating at normal pressure (1-2 atm) for the hydrogenation step significantly lowers the safety risks associated with high-pressure hydrogen gas, simplifying the regulatory compliance burden for manufacturing plants. The use of common organic solvents allows for established recycling and recovery protocols, minimizing the environmental footprint of the process. The straightforward workup procedure, involving filtration and concentration, reduces the volume of aqueous waste generated compared to acid/base extraction methods, aligning with modern green chemistry principles and sustainability goals.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of this bivalent DNJ derivative. These answers are derived directly from the experimental data and technical specifications provided in the patent documentation. Understanding these details is crucial for R&D teams evaluating the feasibility of this molecule for drug development programs and for procurement teams assessing the vendor qualification process. The clarity of the synthetic route and the robustness of the analytical data provide a strong foundation for scaling this technology to commercial levels.
Q: How does the bivalent structure improve alpha-glucosidase inhibition compared to monomers?
A: According to multivalent binding theory, the bivalent structure allows simultaneous binding to multiple receptor sites, significantly enhancing physiological activity and duration compared to monomeric DNJ.
Q: Is the hydrogenation step safe for large-scale manufacturing?
A: Yes, the process utilizes normal pressure hydrogenation (1-2 atm) with 10% Pd/C, which eliminates the need for high-pressure equipment, thereby reducing capital expenditure and safety risks.
Q: What is the expected purity profile of the final intermediate?
A: The method employs standard column chromatography and filtration steps, allowing for the removal of benzyl byproducts and catalyst residues to meet stringent pharmaceutical purity specifications.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Bivalent DNJ Derivative Supplier
At NINGBO INNO PHARMCHEM, we recognize the immense potential of multivalent enzyme inhibitors in the next generation of metabolic disease therapeutics. Our team of expert process chemists has extensively analyzed the synthetic pathway described in CN112441963B and is fully prepared to support your development needs. We possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your transition from benchtop discovery to clinical supply is seamless. Our facilities are equipped with state-of-the-art hydrogenation reactors and rigorous QC labs capable of meeting stringent purity specifications required for API intermediates, guaranteeing consistent quality batch after batch.
We invite you to collaborate with us to optimize this synthesis for your specific requirements. Our technical procurement team is ready to provide a Customized Cost-Saving Analysis tailored to your volume needs, demonstrating how our efficient manufacturing processes can lower your overall project costs. Please contact us today to request specific COA data for our reference standards and to discuss route feasibility assessments for your upcoming projects. Let us be your partner in bringing this promising bivalent DNJ derivative from the laboratory to the market.