Advanced Dehydro Amino Acid Derivatives for Enhanced Polypeptide Drug Stability and Commercial Scalability
Advanced Dehydro Amino Acid Derivatives for Enhanced Polypeptide Drug Stability and Commercial Scalability
The pharmaceutical industry is constantly seeking robust solutions to overcome the inherent instability of polypeptide and protein therapeutics, which are often rapidly degraded by proteases in the human body. Addressing this critical challenge, patent CN110642753A introduces a novel class of amino acid derivatives specifically engineered to modify polypeptide drugs, thereby resisting enzymatic degradation and extending drug half-life. These derivatives, characterized by a unique dehydro-structure, offer a strategic advantage for biopharmaceutical developers aiming to improve the pharmacokinetic profiles of their pipeline candidates. The structural versatility of these compounds allows for precise integration into complex peptide sequences without compromising biological activity.
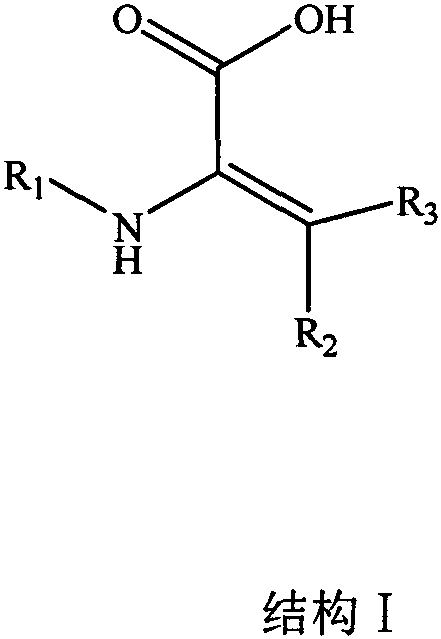
For R&D directors and process chemists, the significance of this technology lies in its dual synthetic approach, providing flexibility depending on the available starting materials. The patent details methods that transform common amino acids into high-value dehydro-variants, creating a reliable pathway for producing stable peptide intermediates. As a leading manufacturer, understanding the nuances of these synthetic routes is essential for evaluating the feasibility of scaling these modifications from laboratory benchtop to commercial production lines.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditionally, enhancing the stability of polypeptide drugs has involved complex PEGylation or the incorporation of non-natural amino acids through laborious solid-phase synthesis protocols that often suffer from low coupling efficiencies. Conventional methods for introducing double bonds into amino acid backbones frequently rely on harsh conditions or expensive chiral catalysts that are difficult to remove, leading to impurity profiles that complicate regulatory approval. Furthermore, standard dehydration reactions often lack regioselectivity, resulting in mixtures of isomers that require extensive and costly purification steps, ultimately driving up the cost of goods sold (COGS) for the final API. The reliance on transition metals in some older methodologies also introduces the risk of heavy metal contamination, necessitating additional downstream processing units that delay time-to-market.
The Novel Approach
The methodology disclosed in patent CN110642753A circumvents these historical bottlenecks by utilizing straightforward organic transformations that are highly amenable to scale-up. By employing a dehydration strategy on hydroxyl-containing amino acids or a dehydrogenation strategy on standard amino acids, the process achieves high selectivity for the desired dehydro-structure. This novel approach eliminates the need for exotic reagents, instead leveraging common protecting group chemistry (such as Fmoc and Benzyl esters) that is already well-established in GMP facilities. The result is a cleaner reaction profile with fewer by-products, simplifying the isolation of the target intermediate and ensuring a higher overall purity suitable for parenteral applications.
Mechanistic Insights into Dehydration and Dehydrogenation Pathways
The core innovation of this technology rests on two distinct mechanistic pathways that allow for the precise installation of the carbon-carbon double bond adjacent to the amino acid alpha-carbon. In the dehydration pathway, the side-chain hydroxyl group is first activated, typically via tosylation, converting a poor leaving group into an excellent one. Subsequent base-mediated elimination facilitates the formation of the double bond with high stereocontrol. Alternatively, the dehydrogenation pathway involves the N-chlorination of the amino group followed by base-induced elimination of HCl. This mechanism is particularly valuable for amino acids lacking side-chain functionality, expanding the scope of accessible derivatives beyond just serine or threonine analogues.
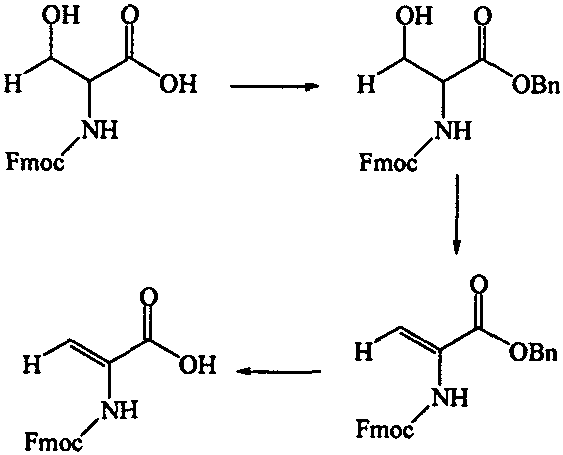
From an impurity control perspective, these mechanisms are advantageous because the intermediates formed are chemically distinct from the starting materials, allowing for easy monitoring via HPLC. For instance, the conversion of Fmoc-Serine benzyl ester to the tosylate intermediate creates a significant shift in polarity, enabling effective separation before the final elimination step. This stepwise control minimizes the formation of racemization by-products, a common concern in amino acid chemistry. The final deprotection steps, such as the use of HBr/TFA, are optimized to cleave protecting groups without hydrogenating the sensitive double bond, preserving the structural integrity required for the derivative's biological function.
How to Synthesize Fmoc-dehydroxyserine Efficiently
The synthesis of Fmoc-dehydroxyserine serves as a prime example of the practical application of this technology, demonstrating a robust three-step sequence that balances yield and operational simplicity. The process begins with the protection of the carboxylic acid, followed by activation of the side chain, and concludes with elimination and deprotection. This route has been validated to provide high yields at each stage, making it an attractive candidate for process optimization. Detailed standardized synthesis steps for this specific transformation are outlined in the guide below, providing a clear roadmap for technical teams looking to replicate these results.
- Protect the carboxyl group of Fmoc-Serine by reacting with benzyl bromide and sodium carbonate in DMF to form Fmoc-serine benzyl ester.
- Activate the side-chain hydroxyl group by reacting with p-toluenesulfonyl chloride in dry pyridine at low temperature (-5°C) to form the tosylate intermediate.
- Perform elimination and deprotection by treating the benzyl ester with HBr/TFA solution, followed by recrystallization to obtain pure Fmoc-dehydroxyserine.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this synthetic technology offers tangible benefits related to raw material security and process economics. The starting materials, such as Fmoc-Serine and Alanine methyl ester, are commodity chemicals produced globally in massive quantities, ensuring a stable and competitive supply base. Unlike processes dependent on proprietary catalysts or rare earth metals, this methodology relies on standard organic reagents like p-toluenesulfonyl chloride and DBU, which are readily available from multiple vendors, mitigating the risk of single-source supply disruptions.
- Cost Reduction in Manufacturing: The elimination of expensive transition metal catalysts and the reduction of purification steps directly contribute to a lower manufacturing cost structure. By achieving high yields in the protection and elimination steps, the overall material throughput is maximized, reducing the waste of valuable chiral starting materials. Furthermore, the use of standard solvents like DMF, pyridine, and ethyl acetate simplifies solvent recovery and recycling operations, leading to substantial operational expenditure savings over the lifecycle of the product.
- Enhanced Supply Chain Reliability: The robustness of the reaction conditions, which tolerate ambient or moderately low temperatures without requiring cryogenic cooling for extended periods, enhances batch consistency and throughput. This operational simplicity translates to shorter cycle times and increased production capacity, allowing suppliers to respond more agilely to fluctuating market demands. The avoidance of hazardous reagents also streamlines logistics and storage requirements, reducing the administrative burden associated with handling controlled substances.
- Scalability and Environmental Compliance: The synthetic routes described are inherently scalable, moving seamlessly from gram-scale laboratory experiments to multi-kilogram pilot runs without significant re-engineering. The waste streams generated are primarily organic salts and solvents, which can be treated using standard effluent management protocols, ensuring compliance with increasingly stringent environmental regulations. This green chemistry profile aligns with the sustainability goals of major pharmaceutical companies, adding value beyond mere cost considerations.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of these amino acid derivatives in drug development programs. These answers are derived directly from the technical specifications and experimental data provided in the patent documentation, ensuring accuracy and relevance for decision-makers. Understanding these details is crucial for assessing the fit of this technology within your specific project timelines and quality requirements.
Q: What is the primary application of these dehydro amino acid derivatives?
A: These derivatives are specifically designed for the modification of polypeptide drugs. By incorporating dehydro-amino acids at enzyme cleavage sites, they effectively block proteolytic degradation, thereby significantly prolonging the half-life and efficacy of the therapeutic protein in vivo.
Q: Are there multiple synthetic routes available for these compounds?
A: Yes, the patent discloses two distinct methods. The first is a dehydration method starting from amino acids containing side-chain hydroxyl groups (like Serine). The second is a dehydrogenation method utilizing amino acids without side-chain hydroxyls (like Alanine) via N-chlorination and elimination.
Q: How does this technology impact supply chain reliability for peptide manufacturers?
A: The synthesis routes utilize widely available starting materials such as Fmoc-Serine and Alanine methyl ester. The processes avoid the use of scarce transition metal catalysts, relying instead on standard organic reagents, which ensures consistent raw material availability and reduces supply chain bottlenecks.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Fmoc-dehydroxyserine Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that high-quality intermediates play in the success of next-generation polypeptide therapeutics. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your transition from clinical trials to market launch is seamless. We maintain stringent purity specifications and operate rigorous QC labs equipped with state-of-the-art analytical instrumentation to guarantee that every batch of Fmoc-dehydroxyserine meets the exacting standards required for GMP manufacturing.
We invite you to engage with our technical procurement team to discuss how our capabilities can support your specific development needs. By requesting a Customized Cost-Saving Analysis, you can gain deeper insights into how our optimized processes can reduce your overall project costs. We encourage potential partners to contact us directly to obtain specific COA data and route feasibility assessments tailored to your unique molecular targets.