Advanced Synthesis of FAK-Targeting Compounds and F-18 Radiolabels for Oncology Applications
Introduction to Next-Generation FAK Targeting Technologies
The landscape of oncology drug development is rapidly evolving, with Focal Adhesion Kinase (FAK) emerging as a pivotal target for both therapeutic intervention and diagnostic imaging. Patent CN111233834A discloses a sophisticated class of small molecule inhibitors designed to disrupt FAK signaling pathways, which are critically involved in tumor metastasis and progression. This intellectual property not only covers potent therapeutic agents but also extends to F-18 radiolabeled analogs, bridging the gap between treatment and early diagnosis through Positron Emission Tomography (PET). For pharmaceutical developers, accessing these advanced chemical entities represents a significant opportunity to enhance pipeline diversity. The disclosed compounds exhibit excellent in vitro inhibitory activity against FAK kinase, with specific embodiments demonstrating IC50 values in the nanomolar range, validating their potential as high-efficacy drug candidates. Furthermore, the inclusion of radiolabeling protocols provides a comprehensive toolkit for theranostic applications, allowing for real-time monitoring of drug distribution and tumor uptake in preclinical models.
From a supply chain perspective, the availability of robust synthetic routes for these complex molecules is paramount. The patent details multiple methodologies for constructing the core pyrimidine-phenyl scaffold, utilizing readily available starting materials such as chloropyrimidines and substituted nitrobenzenes. This strategic design choice mitigates supply risks associated with exotic reagents, ensuring a more stable procurement environment for downstream manufacturers. As a reliable pharmaceutical intermediates supplier, understanding the nuances of these synthetic pathways allows us to anticipate raw material requirements and optimize inventory management. The dual utility of these compounds—as both growth inhibitors and imaging agents—further amplifies their commercial value, offering clients a versatile platform for developing integrated oncology solutions. By leveraging the technical insights from this patent, stakeholders can accelerate the translation of FAK-targeting therapies from bench to bedside.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditional approaches to synthesizing kinase inhibitors often suffer from inefficiencies that hinder commercial viability. Many legacy routes rely on late-stage functionalization strategies that result in poor overall yields and difficult purification profiles. For instance, conventional methods for introducing fluorine atoms or other halogens onto aromatic systems frequently require harsh conditions that compromise the integrity of sensitive functional groups elsewhere in the molecule. Additionally, older radiolabeling techniques often struggle with low specific activity and poor radiochemical purity, necessitating complex and time-consuming HPLC separations that reduce the final usable dose. These bottlenecks not only increase the cost of goods sold (COGS) but also extend the lead time for high-purity pharmaceutical intermediates, creating friction in the development timeline. Furthermore, the lack of modularity in traditional scaffolds limits the ability to rapidly generate analogs for structure-activity relationship (SAR) studies, slowing down the optimization process required to achieve clinical-grade potency.
The Novel Approach
In contrast, the methodology outlined in CN111233834A introduces a modular and convergent synthesis strategy that addresses these historical pain points. The novel approach utilizes a stepwise assembly of the pyrimidine core, allowing for the independent optimization of different molecular regions before final coupling. By employing specific leaving groups such as chlorine or bromine at strategic positions on the pyrimidine ring, the patent enables highly selective nucleophilic aromatic substitution reactions under controlled thermal conditions. This precision minimizes the formation of regioisomers and byproducts, significantly streamlining the purification process. Moreover, the radiolabeling protocol employs a Kryptofix 2.2.2. mediated nucleophilic fluorination, a gold-standard technique known for its efficiency and reproducibility. This ensures that the resulting F-18 labeled compounds maintain high radiochemical purity (>98%) without extensive degradation. The ability to synthesize these compounds with consistent quality and improved yield represents a paradigm shift in cost reduction in oncology drug manufacturing, providing a clear competitive advantage for partners adopting this technology.
Mechanistic Insights into Nucleophilic Substitution and Radiolabeling
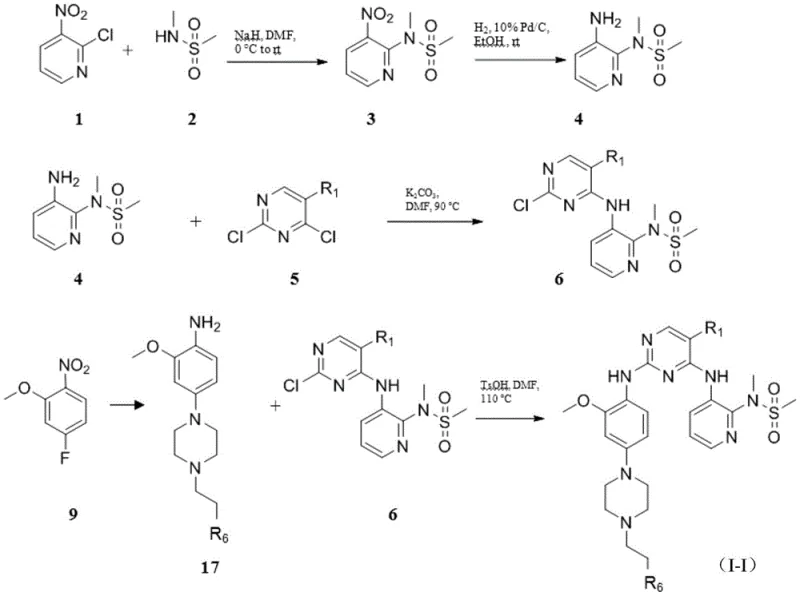
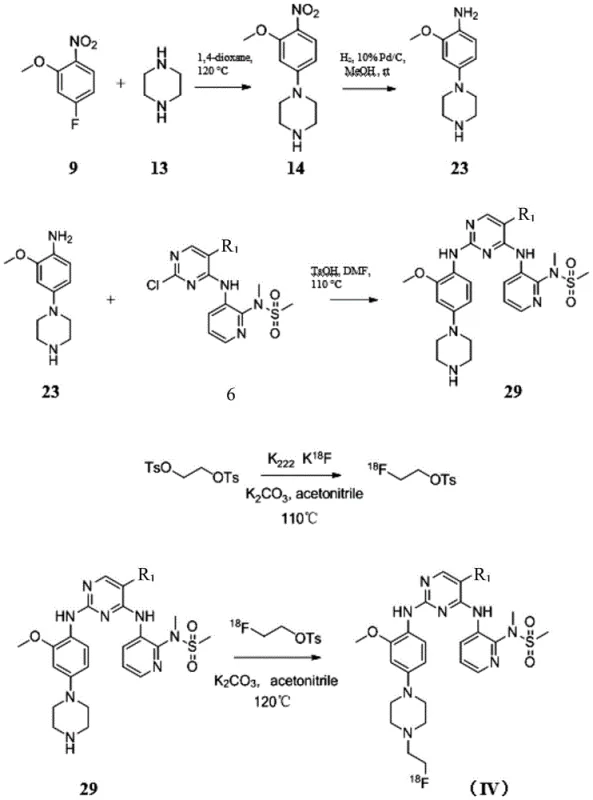
The core chemical transformation driving the synthesis of these FAK inhibitors is the nucleophilic aromatic substitution (SNAr) on the electron-deficient pyrimidine ring. This reaction is facilitated by the presence of electron-withdrawing nitrogen atoms within the heterocycle, which activate the carbon-halogen bonds towards attack by amine nucleophiles. In the described embodiments, amines derived from reduced nitrobenzene precursors act as the nucleophiles, displacing chloride or bromide leaving groups to form the critical C-N bonds that link the pharmacophores. The reaction kinetics are carefully managed through temperature control, typically ranging from 80°C to 120°C, and the use of bases like potassium carbonate or triethylamine to scavenge the generated acid. This mechanistic understanding is crucial for R&D teams aiming to replicate or scale the process, as slight deviations in pH or temperature can lead to hydrolysis of the pyrimidine ring or incomplete conversion. The patent further elucidates the importance of solvent selection, with polar aprotic solvents like DMF or acetonitrile proving optimal for solubilizing both organic substrates and inorganic bases.
Regarding the radiolabeling mechanism, the process relies on the high reactivity of the [18F]fluoride ion when complexed with Kryptofix 2.2.2. and potassium carbonate. This complex enhances the nucleophilicity of the fluoride anion in organic solvents, enabling it to displace good leaving groups such as tosylates or mesylates on the aliphatic side chains of the precursor molecules. The reaction is typically conducted at elevated temperatures (around 110°C to 120°C) to overcome the activation energy barrier within a short timeframe, which is essential given the short half-life of Fluorine-18. Following the labeling event, rigorous purification via semi-preparative HPLC is employed to separate the desired radiotracer from unreacted precursor and side products. This step is critical for ensuring that the final injectable solution meets stringent regulatory standards for chemical and radiochemical purity. Understanding these mechanistic details allows for better troubleshooting during scale-up and ensures that impurity profiles remain within acceptable limits for clinical applications.
How to Synthesize FAK Inhibitor Intermediates Efficiently
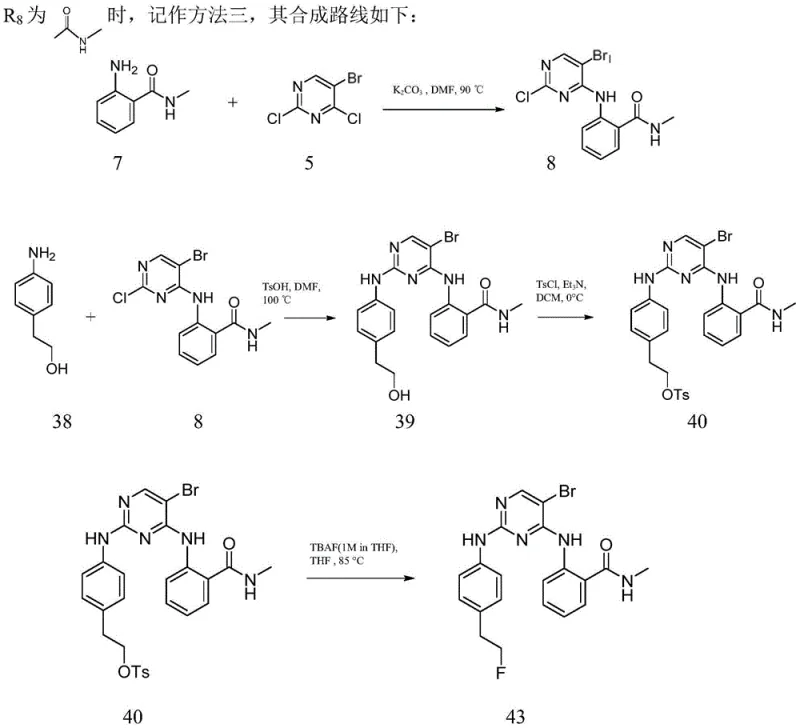
The synthesis of these high-value intermediates requires a disciplined approach to reaction engineering and process control. The patent outlines a series of interconnected steps that transform simple commodity chemicals into complex bioactive molecules. Key to this process is the preparation of the amine intermediates via catalytic hydrogenation, which must be performed under strict safety protocols due to the use of pressurized hydrogen gas. Following the reduction, the coupling reactions demand precise stoichiometric control to prevent over-alkylation or polymerization. Detailed standardized synthesis steps are essential for maintaining batch-to-batch consistency, particularly when transitioning from gram-scale laboratory experiments to kilogram-scale production. The following guide summarizes the critical operational parameters derived from the patent examples, serving as a foundational reference for process chemists.
- Perform nucleophilic substitution on chloropyrimidine scaffolds using specific amine intermediates under basic conditions at elevated temperatures.
- Execute catalytic hydrogenation of nitro-groups to amines using Pd/C catalysts under pressurized hydrogen environments.
- Conduct final coupling reactions with p-toluenesulfonic acid catalysis followed by purification via column chromatography or HPLC.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain leaders, the adoption of the synthetic routes described in this patent offers tangible strategic benefits beyond mere technical feasibility. The reliance on commercially available starting materials, such as 2-chloro-3-nitropyridine and various substituted benzoic acids, significantly de-risks the supply chain. Unlike proprietary reagents that may be sourced from a single vendor, these bulk chemicals are produced by multiple global suppliers, fostering a competitive pricing environment and ensuring continuity of supply even during market disruptions. This diversification of the raw material base is a critical factor in enhancing supply chain reliability, allowing manufacturers to negotiate better terms and avoid single-source bottlenecks. Furthermore, the synthetic steps utilize standard unit operations like filtration, extraction, and distillation, which can be easily accommodated in existing multipurpose manufacturing facilities without requiring specialized capital investment.
- Cost Reduction in Manufacturing: The streamlined synthesis pathway minimizes the number of isolation steps and reduces the consumption of expensive catalysts and solvents. By achieving high conversion rates in the key coupling reactions, the process lowers the overall material input required per kilogram of final product. Additionally, the avoidance of precious metal catalysts in certain steps, favoring more abundant alternatives where possible, contributes to substantial cost savings. The high purity of the intermediates generated reduces the burden on downstream purification, thereby lowering waste disposal costs and improving overall process mass intensity. These efficiencies collectively drive down the cost of goods, making the final therapeutic or diagnostic agent more economically viable for healthcare systems.
- Enhanced Supply Chain Reliability: The robustness of the chemical transformations described ensures consistent output quality, which is vital for maintaining regulatory compliance and avoiding production delays. The use of stable intermediates that can be stored for extended periods allows for the decoupling of upstream and downstream manufacturing stages. This flexibility enables companies to build strategic inventory buffers of key precursors, mitigating the impact of unexpected demand surges or logistical challenges. Moreover, the scalability of the reactions means that production volumes can be ramped up quickly to meet clinical trial requirements or commercial launch demands without the need for extensive process re-validation.
- Scalability and Environmental Compliance: The synthetic routes are designed with green chemistry principles in mind, utilizing solvents that are easier to recover and recycle. The high atom economy of the nucleophilic substitution reactions minimizes the generation of hazardous byproducts, simplifying waste treatment protocols. As production scales from pilot plants to commercial facilities, these environmental advantages translate into lower operational costs related to effluent treatment and regulatory reporting. The ability to execute these syntheses safely and sustainably at large volumes positions partners as responsible stewards of chemical manufacturing, aligning with the increasing ESG (Environmental, Social, and Governance) expectations of global pharmaceutical clients.
Frequently Asked Questions (FAQ)
The technical complexity of FAK inhibitor synthesis often raises questions regarding process optimization, regulatory alignment, and practical implementation. Stakeholders frequently seek clarification on the specific conditions required to achieve the reported yields and purity levels, as well as the compatibility of these methods with existing GMP (Good Manufacturing Practice) infrastructure. Addressing these inquiries proactively helps to build confidence in the technology and facilitates smoother technology transfer processes. The following answers are derived directly from the experimental data and procedural descriptions found within the patent documentation, providing authoritative guidance for technical teams evaluating this opportunity.
Q: What represents the key structural advantage of these FAK inhibitors?
A: The compounds feature a pyrimidine core linked to substituted phenyl rings, allowing for versatile modification at the R1 and R4 positions to optimize kinase binding affinity and metabolic stability.
Q: How is the F-18 radiolabeling achieved in this protocol?
A: Radiolabeling is accomplished via nucleophilic substitution using Kryptofix 2.2.2./[18F]KF/K2CO3 complexes on tosylate precursors, ensuring high radiochemical purity suitable for PET imaging.
Q: Are these intermediates scalable for commercial production?
A: Yes, the synthesis relies on standard organic transformations such as SNAr and hydrogenation which are well-established for scale-up from laboratory to multi-ton commercial manufacturing.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable FAK Inhibitor Intermediates Supplier
At NINGBO INNO PHARMCHEM, we recognize the transformative potential of FAK-targeting technologies in the fight against cancer. Our team of expert process chemists has thoroughly analyzed the synthetic routes disclosed in CN111233834A and is fully prepared to support your development goals. We possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your transition from preclinical research to clinical supply is seamless and efficient. Our state-of-the-art facilities are equipped with the necessary reactors and purification systems to handle the specific thermal and pressure requirements of these syntheses, while our rigorous QC labs guarantee that every batch meets stringent purity specifications. We understand that time-to-market is critical in oncology, and our agile project management approach is designed to accelerate your timelines without compromising on quality or safety.
We invite you to engage with our technical procurement team to discuss how we can tailor our manufacturing capabilities to your specific needs. Whether you require a Customized Cost-Saving Analysis for your current supply chain or need to verify the feasibility of scaling a specific analog, we are here to provide the data-driven insights you need. Please contact us to request specific COA data for our catalog intermediates or to initiate a conversation about route feasibility assessments for your proprietary candidates. Together, we can advance the next generation of FAK inhibitors and radiopharmaceuticals, bringing life-saving diagnostics and therapies to patients faster and more affordably.