Advanced Commercial Synthesis of Ibrutinib via Novel Mitsunobu Reagent and Protection Strategy
The pharmaceutical industry's demand for high-purity Bruton's tyrosine kinase (BTK) inhibitors continues to surge, driven by the critical role of Ibrutinib in treating various B-cell malignancies. Patent CN116063309B introduces a transformative synthesis method that addresses longstanding challenges in impurity control and yield optimization. This technical breakthrough centers on a strategic BOC protection protocol and the deployment of a novel Mitsunobu reagent, fundamentally altering the efficiency landscape for reliable pharmaceutical intermediate supplier networks. By mitigating the formation of complex byproducts that typically plague conventional routes, this methodology ensures a robust supply chain for high-purity Ibrutinib. The structural integrity of the final product, as depicted below, is maintained through rigorous process controls that eliminate thermodynamic instability issues found in earlier iterations.
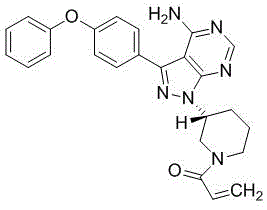
Furthermore, the integration of mild reaction conditions ranging from 20°C to 30°C significantly lowers the energy footprint and safety risks associated with high-temperature cyclization steps seen in prior art. This approach not only enhances operator safety but also simplifies the engineering requirements for reactor systems, facilitating smoother technology transfer from laboratory to pilot plant scales. For procurement teams, the shift away from hazardous reagents like trimethylsilyldiazomethane represents a substantial reduction in regulatory compliance burdens and waste disposal costs. Consequently, this patent provides a viable pathway for cost reduction in API manufacturing while adhering to stringent environmental, health, and safety (EHS) standards required by global regulatory bodies.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of Ibrutinib has been fraught with chemical inefficiencies, particularly regarding the management of the reactive amino group on the pyrimidine ring. As illustrated in comparative routes such as those disclosed in CN101610676, the absence of a protecting group during the Mitsunobu reaction leads to the generation of significant impurity profiles. Specifically, the unprotected amino group acts as a competing nucleophile, resulting in the formation of bis-amidation impurities and dimerization byproducts that are structurally similar to the target molecule. These impurities, often designated as Formula II, III, and IV in technical literature, possess polarities and solubilities that closely mimic the active pharmaceutical ingredient, rendering their removal via standard crystallization or chromatography exceptionally difficult and costly.
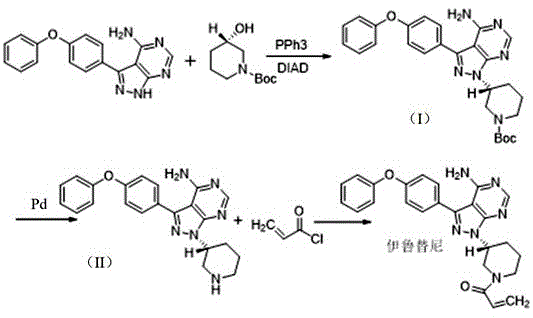
Moreover, traditional methodologies frequently rely on aggressive reagents and extreme thermal conditions to drive conversions, which exacerbates the degradation of sensitive intermediates. For instance, certain legacy routes require pyrimidine cyclization temperatures exceeding 180°C, creating unfavorable thermodynamics for industrial scalability and increasing the risk of thermal runaway events. The reliance on expensive supported triphenylphosphine catalysts in Mitsunobu reactions further inflates the raw material costs, while the use of explosive substances poses severe logistical challenges for storage and transportation. These cumulative factors result in low overall yields, often hovering around 30-40%, which directly impacts the economic viability of commercial scale-up of complex kinase inhibitors and creates bottlenecks in the global supply chain.
The Novel Approach
The innovative strategy outlined in Patent CN116063309B effectively circumvents these pitfalls through a preemptive protection-deprotection sequence that isolates the reactive amino functionality. By employing di-tert-butyl dicarbonate to install a BOC group prior to the coupling step, the synthesis effectively masks the nucleophilicity of the pyrimidine nitrogen, thereby preventing the formation of bis-amidation side products during the subsequent acylation with acryloyl chloride. This strategic modification ensures that the acylation reaction proceeds with high regioselectivity exclusively at the piperidine nitrogen, drastically simplifying the downstream purification process. The result is a final product with purity levels reaching 99.92%, achieved through straightforward recrystallization rather than resource-intensive preparative HPLC.
In addition to protection chemistry, the process introduces a proprietary Mitsunobu reagent, 1,1'-(azodicarbonyl)ditetrahydroindole, which serves as a superior alternative to conventional azodicarboxylates. This specialized reagent enhances the electrophilicity of the reaction intermediate, facilitating a more efficient displacement of the hydroxyl group on the piperidine ring. The implementation of this reagent has been shown to elevate the yield of the critical coupling step from a mediocre 40-50% range to an impressive >65%, representing a quantum leap in process efficiency. Such improvements are critical for reducing lead time for high-purity pharmaceutical intermediates, as higher yields per batch reduce the number of cycles required to meet production quotas, thereby optimizing facility utilization rates.
Mechanistic Insights into Specialized Mitsunobu Coupling
The core mechanistic advantage of this synthesis lies in the electronic properties of the novel azo reagent, 1,1'-(azodicarbonyl)ditetrahydroindole. Unlike traditional reagents such as DEAD or DIAD, which feature electron-withdrawing alkoxy groups, this custom molecule incorporates electron-donating alkyl-substituted amino groups within a tetrahydroindole framework. This structural variation significantly enhances the basicity and nucleophilicity of the azo nitrogen atoms, allowing for more effective proton abstraction from the substrate alcohol. The resulting betaine intermediate is more stable and reactive, promoting a faster and more complete inversion of configuration at the chiral center of the piperidine ring. This mechanistic refinement is crucial for maintaining the stereochemical integrity of the (3R)-piperidine moiety, which is essential for the biological activity of the final BTK inhibitor.
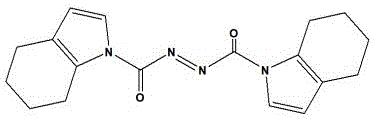
Furthermore, the protection strategy fundamentally alters the reaction landscape by eliminating competitive pathways. In the absence of the BOC group, the primary amine on the pyrimidine ring can attack the activated phosphonium intermediate, leading to N-alkylation side products that are irreversible under the reaction conditions. By sterically and electronically blocking this site, the reaction trajectory is forced exclusively towards the desired O-alkylation of the piperidine hydroxyl group. This selectivity is further reinforced during the final acylation step, where the protected amine remains inert to acryloyl chloride, preventing the formation of the thermodynamically stable bis-amidine impurities observed in unprotected routes. The comprehensive reaction scheme below details the seamless integration of these protective and coupling strategies into a cohesive five-step workflow.
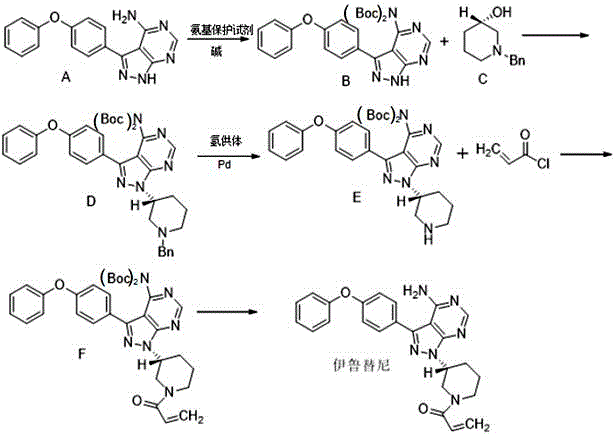
The deprotection phases are equally optimized for industrial practicality, utilizing catalytic hydrogenation with ammonium formate as a hydrogen donor. This transfer hydrogenation method avoids the need for high-pressure hydrogen gas equipment, reducing capital expenditure and safety risks. The final removal of the BOC group using trichloroacetic acid or trifluoroacetic acid proceeds under mild acidic conditions that do not compromise the integrity of the acrylamide double bond, ensuring the final API retains its full potency. This holistic approach to mechanism design demonstrates a deep understanding of physical organic chemistry, translating theoretical advantages into tangible manufacturing benefits such as reduced solvent consumption and minimized waste generation.
How to Synthesize Ibrutinib Efficiently
The execution of this advanced synthesis requires precise control over stoichiometry and temperature to maximize the benefits of the novel reagents and protection groups. The process begins with the careful preparation of the BOC-protected pyrimidine intermediate, followed by the critical Mitsunobu coupling where the custom azo reagent is introduced. Subsequent steps involve catalytic deprotection, selective acylation, and final acid hydrolysis, each designed to maintain high purity without the need for complex chromatographic separations. Detailed operational parameters, including specific molar ratios and solvent systems, are essential for replicating the high yields reported in the patent data.
- Protect the amino group on the pyrimidine ring of 3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine using di-tert-butyl dicarbonate to prevent side reactions.
- Perform the Mitsunobu reaction using the novel reagent 1,1'-(azodicarbonyl)ditetrahydroindole to couple the protected intermediate with (S)-1-benzyl-3-hydroxypiperidine, significantly boosting yield.
- Execute catalytic hydrogenation for benzyl deprotection, followed by acylation with acryloyl chloride and final acid-mediated removal of the BOC group to obtain the target API.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this synthesis route offers compelling economic and logistical advantages that extend beyond simple yield metrics. The elimination of hazardous and explosive reagents such as trimethylsilyldiazomethane significantly lowers the barrier for entry for contract manufacturing organizations (CMOs), expanding the pool of qualified suppliers and enhancing supply chain resilience. Additionally, the use of readily available and cost-effective starting materials, combined with the high efficiency of the novel Mitsunobu reagent, drives down the cost of goods sold (COGS). The ability to achieve high purity through simple crystallization rather than expensive purification technologies translates directly into margin improvement and price competitiveness in the generic API market.
- Cost Reduction in Manufacturing: The significant increase in reaction yield, particularly in the coupling step, reduces the amount of raw materials required per kilogram of finished product. By avoiding the formation of difficult-to-remove impurities, the process eliminates the need for costly and time-consuming preparative chromatography, leading to substantial savings in solvent usage and labor hours. The replacement of expensive supported catalysts with standard triphenylphosphine further optimizes the raw material bill, ensuring a more favorable economic model for large-scale production.
- Enhanced Supply Chain Reliability: The reliance on stable, non-explosive reagents simplifies logistics and storage requirements, reducing the risk of supply disruptions caused by regulatory restrictions on hazardous chemicals. The mild reaction conditions allow for the use of standard glass-lined or stainless steel reactors, which are widely available in the global CDMO network, ensuring that production capacity can be easily scaled up to meet market demand without requiring specialized infrastructure investments.
- Scalability and Environmental Compliance: The process generates fewer byproducts and utilizes safer solvents, aligning with green chemistry principles and reducing the environmental burden of waste treatment. The high selectivity of the reaction minimizes the generation of organic waste streams, lowering disposal costs and facilitating compliance with increasingly stringent environmental regulations. This sustainability profile makes the route attractive for long-term partnerships with pharmaceutical companies committed to reducing their carbon footprint.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis method. These insights are derived directly from the experimental data and comparative analysis provided in the patent documentation, offering clarity on the practical implications of adopting this technology for commercial manufacturing.
Q: Why is amino protection critical in Ibrutinib synthesis?
A: Without protection, the primary amino group on the pyrimidine ring participates in unwanted side reactions during the Mitsunobu coupling and subsequent acylation, leading to difficult-to-remove bis-amidation impurities that compromise final purity.
Q: What is the advantage of using 1,1'-(azodicarbonyl)ditetrahydroindole?
A: This custom reagent replaces traditional azodicarboxylates like DEAD or DIAD, offering superior electron-donating capabilities that enhance proton combination with the substrate, thereby increasing the reaction yield from approximately 40-50% to over 65%.
Q: Is this synthesis route suitable for large-scale production?
A: Yes, the process utilizes mild reaction temperatures (20-30°C), avoids explosive reagents like trimethylsilyldiazomethane, and achieves high purity (99.92%) through simple crystallization, making it highly scalable and safe for industrial manufacturing.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Ibrutinib Supplier
At NINGBO INNO PHARMCHEM, we leverage our extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production to bring this advanced synthesis method to life. Our state-of-the-art facilities are equipped to handle the specific requirements of this process, including the safe handling of specialized reagents and the precise temperature control needed for optimal yield. We adhere to stringent purity specifications and operate rigorous QC labs to ensure that every batch of Ibrutinib meets the highest international standards for oncology therapeutics. Our commitment to quality assurance guarantees a consistent supply of high-performance intermediates and APIs for your drug development pipelines.
We invite you to engage with our technical procurement team to discuss how this optimized route can benefit your specific project needs. By requesting a Customized Cost-Saving Analysis, you can gain a deeper understanding of the potential economic impact of switching to this superior synthesis method. We encourage you to contact us today to obtain specific COA data and route feasibility assessments tailored to your volume requirements, ensuring a seamless transition to a more efficient and reliable supply source.