Scalable Synthesis of Cyclic Pentapeptide Intermediates for Advanced Antitumor Drug Development
The pharmaceutical industry is constantly seeking robust pathways to access complex cyclic peptides, which serve as potent scaffolds for next-generation therapeutics. A pivotal advancement in this domain is documented in patent CN102329376A, which discloses a highly efficient synthesis method for Cyclo(phenylalanyl-N-methylleucyl-leucyl-N-methylleucyl-leucyl), a novel cyclic pentapeptide derivative of Galaxamide. This compound has demonstrated significant cytotoxic activity against various human cancer cell lines, including liver, breast, and cervical cancer, positioning it as a valuable lead compound for antitumor drug development. The disclosed methodology overcomes traditional bottlenecks in peptide macrocyclization by employing a strategic '2+3' convergent approach, ensuring both high stereochemical fidelity and improved overall yields. For R&D teams and procurement specialists alike, understanding this route provides a critical blueprint for sourcing high-purity intermediates essential for preclinical and clinical trials.
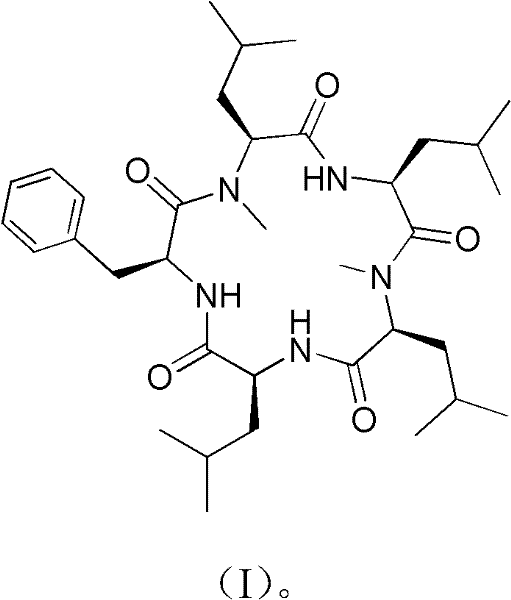
Furthermore, the technical elegance of this synthesis lies in its ability to navigate the steric challenges posed by N-methylated amino acids. By carefully selecting the ring-closing position between the amino group of phenylalanine and the carboxyl group of leucine, the process effectively avoids the difficult ortho-cyclization adjacent to N-methyl groups. This strategic decision not only simplifies the reaction profile but also enhances the feasibility of large-scale manufacturing. As a reliable pharmaceutical intermediate supplier, recognizing the nuances of such patented processes allows us to offer clients not just a molecule, but a validated, scalable supply chain solution that mitigates the risks associated with complex peptide synthesis.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditional linear synthesis of cyclic peptides often suffers from diminishing returns as the chain length increases, primarily due to the accumulation of side reactions and purification losses at each coupling step. In conventional approaches, attempting to cyclize a linear pentapeptide containing multiple N-methylated residues frequently results in low yields and significant epimerization, rendering the process economically unviable for commercial production. The steric bulk introduced by N-methyl groups creates substantial kinetic barriers during the final macrocyclization step, often requiring harsh conditions that degrade the product or generate difficult-to-remove impurities. Moreover, linear strategies typically lack the modularity needed to quickly optimize specific segments of the peptide, making process development slow and resource-intensive. These limitations have historically constrained the availability of high-quality cyclic peptide intermediates, forcing research teams to rely on small-batch, high-cost supplies that hinder rapid drug discovery progress.
The Novel Approach
In stark contrast, the methodology outlined in CN102329376A introduces a sophisticated '2+3' convergent synthesis strategy that fundamentally reshapes the efficiency landscape. By independently synthesizing a dipeptide fragment and a tripeptide fragment, the process isolates complexity and allows for rigorous quality control of each segment before the final assembly. This modular approach drastically reduces the number of sequential steps required for the final chain elongation, thereby preserving the overall yield. The innovation extends to the specific choice of cyclization reagents and conditions; the use of a mixed solvent system comprising tetrahydrofuran, dichloromethane, and acetonitrile, combined with potent coupling agents like HATU and TBTU, facilitates a smooth ring closure even in the presence of sterically demanding N-methyl groups. This novel pathway transforms a previously challenging synthesis into a robust, reproducible protocol suitable for industrial application, offering a clear path toward cost reduction in API manufacturing for antitumor agents.
Mechanistic Insights into Convergent Peptide Condensation
The core of this synthesis relies on the precise activation of carboxylic acids using phosphonium and uronium-based coupling reagents, specifically DEPBT, HATU, and TBTU. During the condensation of the dipeptide and tripeptide fragments, DEPBT acts as a highly efficient activating agent that forms an active ester intermediate with minimal risk of racemization. The reaction mechanism involves the nucleophilic attack of the free amine on the activated carbonyl carbon, facilitated by the base DIPEA, which scavenges the generated acid and maintains the optimal pH for coupling. This careful control of reaction kinetics is paramount when dealing with N-methylated amino acids, as their reduced nucleophilicity requires more potent activation to drive the reaction to completion without resorting to excessive heat that could compromise stereochemistry. The subsequent macrocyclization step leverages the synergistic effect of multiple coupling reagents in a polar aprotic environment, ensuring that the entropic penalty of ring closure is overcome effectively.
Impurity control is meticulously managed through a sequence of orthogonal protection and deprotection strategies. The use of tert-butyloxycarbonyl (Boc) groups for N-terminal protection and benzyl esters for C-terminal protection allows for selective removal under distinct conditions—acidolysis for Boc and hydrogenolysis for benzyl esters. This orthogonality ensures that intermediate fragments can be purified to homogeneity before the final assembly, preventing the propagation of impurities into the final cyclic product. For instance, the removal of the benzyl group via catalytic hydrogenation using 10% Pd/C is performed under mild conditions (20-30°C), preserving the integrity of the peptide backbone while cleanly exposing the carboxylic acid needed for cyclization. Such rigorous attention to mechanistic detail ensures that the final product meets the stringent purity specifications demanded by regulatory bodies for pharmaceutical intermediates.
How to Synthesize Cyclo(Phe-N-Me-Leu-Leu-N-Me-Leu-Leu) Efficiently
The synthesis of this complex cyclic pentapeptide is achieved through a multi-step sequence that prioritizes yield and purity at every stage. The process begins with the protection and N-methylation of starting amino acids, followed by the sequential assembly of dipeptide and tripeptide fragments using advanced coupling chemistry. The final stages involve the convergence of these fragments into a linear pentapeptide, followed by global deprotection and macrocyclization to form the target ring structure. This streamlined workflow minimizes handling time and maximizes material throughput, making it an ideal candidate for technology transfer. The detailed standardized synthesis steps, including specific molar ratios, solvent volumes, and reaction times, are provided in the guide below to ensure reproducibility.
- Perform N-terminal protection of L-leucine and L-phenylalanine using Boc2O, followed by N-methylation of leucine derivatives using methyl iodide and sodium hydride.
- Synthesize the dipeptide and tripeptide fragments separately using DEPBT/DIPEA coupling agents, ensuring high stereochemical integrity.
- Condense the protected linear pentapeptide fragments, remove terminal protecting groups via hydrogenation and acid treatment, and execute final macrocyclization using HATU/TBTU reagents.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this patented synthesis route offers profound strategic advantages beyond mere technical feasibility. The reliance on abundant, commodity-grade raw materials such as L-leucine and L-phenylalanine decouples production from the volatility of exotic reagent markets, ensuring a stable and predictable supply base. Furthermore, the mild reaction conditions, predominantly operating between 0°C and 30°C, significantly reduce energy consumption and eliminate the need for specialized high-temperature or high-pressure equipment. This operational simplicity translates directly into lower capital expenditure and reduced operational costs, allowing for more competitive pricing structures without compromising on quality. The robustness of the process also implies a lower risk of batch failures, enhancing supply continuity for critical drug development programs.
- Cost Reduction in Manufacturing: The elimination of expensive transition metal catalysts in the coupling steps, replaced by efficient organic reagents like DEPBT, removes the need for costly and time-consuming heavy metal clearance procedures. This simplification of the downstream processing workflow leads to substantial cost savings in purification and waste management. Additionally, the high yields achieved in key steps, such as the 96.2% yield in the initial protection phase and the 56% yield in the challenging cyclization step, maximize the utilization of raw materials. By optimizing atom economy and reducing the number of purification cycles required, the overall cost of goods sold is significantly driven down, providing a compelling economic case for large-scale production.
- Enhanced Supply Chain Reliability: The modular nature of the '2+3' synthesis strategy allows for the parallel production of dipeptide and tripeptide intermediates, effectively shortening the critical path for manufacturing. This parallelization capability means that bottlenecks in one segment do not necessarily halt the entire production line, thereby improving overall throughput and reducing lead times for high-purity pharmaceutical intermediates. Moreover, the use of standard solvents and reagents that are globally sourced ensures that supply chain disruptions are minimized. The ability to source precursors from multiple qualified vendors further strengthens supply security, making this route highly resilient against market fluctuations and geopolitical instabilities.
- Scalability and Environmental Compliance: The process is designed with scalability in mind, utilizing reaction conditions that are easily transferable from laboratory glassware to industrial reactors. The absence of extreme temperatures or pressures simplifies the engineering requirements for scale-up, reducing the time and cost associated with process validation. From an environmental perspective, the use of recyclable solvents and the generation of benign byproducts align with modern green chemistry principles. The efficient use of reagents minimizes waste generation, lowering the burden on waste treatment facilities and ensuring compliance with increasingly stringent environmental regulations. This sustainable approach not only mitigates regulatory risk but also enhances the corporate social responsibility profile of the manufacturing operation.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the synthesis and application of this cyclic pentapeptide. These insights are derived directly from the experimental data and beneficial effects described in the patent literature, providing a transparent view of the technology's capabilities. Understanding these details is crucial for stakeholders evaluating the feasibility of incorporating this intermediate into their drug development pipelines.
Q: What is the key advantage of the 2+3 synthesis strategy for this cyclic pentapeptide?
A: The 2+3 convergent strategy significantly improves overall yield compared to linear synthesis by minimizing cumulative losses. Crucially, selecting the cyclization site between the phenylalanine amino group and leucine carboxyl group avoids steric hindrance associated with N-methylated residues, leading to a superior cyclization yield of 56%.
Q: How does this process ensure high purity for pharmaceutical applications?
A: The process utilizes robust purification methods at each stage, including silica gel column chromatography and reverse-phase HPLC for the final product. The use of specific coupling reagents like DEPBT minimizes racemization, ensuring the final cyclic peptide meets stringent purity specifications required for antitumor drug candidates.
Q: Are the raw materials for this synthesis readily available for scale-up?
A: Yes, the synthesis relies on commodity amino acids such as L-leucine and L-phenylalanine, which are abundant and cost-effective. The reagents used, including Boc anhydride and standard coupling agents, are commercially available in bulk quantities, facilitating seamless transition from laboratory to industrial production.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Cyclo(Phe-N-Me-Leu-Leu-N-Me-Leu-Leu) Supplier
The technical potential of Cyclo(phenylalanyl-N-methylleucyl-leucyl-N-methylleucyl-leucyl) as a potent antitumor agent is undeniable, yet realizing its commercial value requires a manufacturing partner with deep expertise in complex peptide chemistry. NINGBO INNO PHARMCHEM stands at the forefront of this field, possessing extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production. Our state-of-the-art facilities are equipped to handle the nuanced requirements of peptide synthesis, including strict temperature control and advanced purification capabilities. We adhere to stringent purity specifications and operate rigorous QC labs to ensure that every batch delivered meets the highest international standards, providing our partners with the confidence needed to advance their clinical programs.
We invite you to engage with our technical procurement team to discuss how we can tailor this synthesis route to your specific volume and quality requirements. By leveraging our process optimization capabilities, we can provide a Customized Cost-Saving Analysis that identifies further efficiencies specific to your supply chain needs. We encourage potential partners to request specific COA data and route feasibility assessments to verify our capabilities firsthand. Together, we can accelerate the development of life-saving antitumor therapies by ensuring a reliable, high-quality supply of this critical pharmaceutical intermediate.