Advanced One-Step Synthesis of P-Aminobenzoic Acid Pseudo-Dipeptide Esters for Diabetes Drug Development
Introduction to Novel Anti-Diabetic Intermediate Synthesis
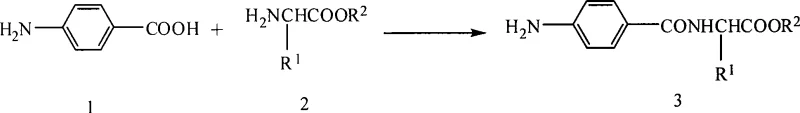
The escalating global prevalence of diabetes has intensified the search for novel therapeutic agents, driving significant innovation in the synthesis of key pharmaceutical intermediates. Patent CN101445468A introduces a groundbreaking methodology for preparing p-aminobenzoic acid pseudo-dipeptide esters, a class of compounds demonstrating promising PPAR agonistic activity. Unlike conventional multi-step syntheses that rely on cumbersome protection-deprotection strategies or hazardous nitro-reduction pathways, this invention utilizes a direct coupling approach between unprotected p-aminobenzoic acid and various amino acid esters. This technical breakthrough not only simplifies the synthetic route but also enhances the purity profile of the final product, addressing critical pain points for R&D teams focused on rapid lead optimization. By leveraging mild reaction conditions and readily available starting materials, this process represents a paradigm shift towards more efficient and sustainable manufacturing of complex peptide-mimetic structures.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of amino acid ester derivatives of substituted p-aminobenzoic acids has been plagued by inefficient multi-step protocols that hinder scalability and increase production costs. The first traditional approach typically involves the use of protected p-aminobenzoic acid, where the amino group must be masked with bulky protecting groups like Boc or Fmoc prior to coupling. This necessitates an additional deprotection step post-coupling, which often requires harsh acidic or basic conditions that can compromise the integrity of sensitive ester functionalities. Alternatively, the second common route starts from p-nitrobenzoic acid, converting it to an acyl chloride before coupling, followed by a reduction of the nitro group. This pathway introduces significant safety hazards associated with acyl chlorides and catalytic hydrogenation or metal-mediated reductions, while also generating substantial waste streams. Both methods suffer from low total yields due to cumulative losses across multiple isolation steps, making them economically unviable for large-scale commercial production of high-purity pharmaceutical intermediates.
The Novel Approach
In a striking departure from established norms, the patented method achieves the direct coupling of unprotected p-aminobenzoic acid with amino acid esters in a single operational step. This innovative strategy eliminates the need for amino protection and subsequent deprotection, as well as the dangerous nitro-reduction sequence, thereby drastically streamlining the workflow. The reaction proceeds smoothly in the presence of specific coupling agents and additives, yielding the target pseudo-dipeptide esters with high efficiency and minimal byproduct formation. By operating under mild temperatures ranging from 10°C to 35°C, the process ensures excellent control over stereochemistry, minimizing the risk of racemization which is a critical quality attribute for chiral drug candidates. This simplified approach not only reduces the consumption of reagents and solvents but also shortens the overall production cycle time, offering a robust solution for the cost reduction in pharmaceutical intermediate manufacturing.
Mechanistic Insights into Carbodiimide-Mediated Direct Coupling
The success of this direct coupling methodology relies heavily on the precise selection of activation reagents that can selectively activate the carboxylic acid of p-aminobenzoic acid without inducing excessive self-polymerization or side reactions with the free aniline nitrogen. The patent highlights the efficacy of carbodiimides such as N,N'-diisopropylcarbodiimide (DIC) or N,N'-dicyclohexylcarbodiimide (DCC) when paired with 1-hydroxybenzotriazole (HOBt). Mechanistically, the carbodiimide reacts with the carboxyl group to form an unstable O-acylisourea intermediate, which is highly susceptible to nucleophilic attack. However, in the absence of additives, this intermediate can rearrange to an unreactive N-acylurea or react with the free amine of another p-aminobenzoic acid molecule. The inclusion of HOBt is crucial as it rapidly converts the O-acylisourea into a more stable and less racemization-prone active ester species. This active ester is sufficiently electrophilic to react with the amino group of the amino acid ester but stable enough to prevent unwanted side reactions, ensuring high chemoselectivity even in the presence of the competing nucleophilic aniline moiety on the benzoic acid ring.
Furthermore, the control of impurities, particularly optical purity, is paramount in the synthesis of chiral intermediates for anti-diabetic drugs. The mild reaction temperature window of 10°C to 35°C specified in the patent plays a vital role in suppressing racemization at the alpha-carbon of the amino acid component. Higher temperatures typically accelerate the rate of oxazolone formation, a key pathway for racemization in peptide coupling, whereas lower temperatures favor the desired amidation kinetics. The patent data indicates that as the reaction temperature decreases within this range, the purity of the product increases, facilitating easier downstream purification. This mechanistic understanding allows process chemists to fine-tune reaction parameters to maximize the yield of the desired enantiomer, ensuring that the final API meets stringent regulatory specifications for chiral purity without the need for expensive chiral resolution steps later in the synthesis.
How to Synthesize P-Aminobenzoic Acid Pseudo-Dipeptide Esters Efficiently
The practical implementation of this synthesis involves a straightforward procedure that can be easily adapted for both laboratory scale-up and industrial production. The process begins with the preparation of the amino acid ester solution, typically generated in situ by treating the corresponding hydrochloride or tosylate salt with a base like triethylamine in tetrahydrofuran (THF). Simultaneously, the p-aminobenzoic acid is activated in a separate vessel using the chosen coupling system. The detailed standardized synthesis steps are provided below to guide technical teams in replicating these high-yielding results.
- Dissolve the amino acid ester salt in THF with triethylamine to generate the free base solution.
- Activate p-aminobenzoic acid in THF using a coupling agent like DIC or DCC alongside HOBt at room temperature.
- Combine the solutions, stir at 10-35°C until completion, and purify the crude product via recrystallization or chromatography.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the transition to this direct coupling technology offers tangible benefits that extend beyond mere chemical elegance. By collapsing a multi-step sequence into a single pot operation, the process inherently reduces the consumption of raw materials, solvents, and energy, leading to substantial cost savings in manufacturing. The elimination of protection and deprotection reagents, which are often expensive and generate significant waste, directly lowers the bill of materials. Moreover, the avoidance of hazardous reagents like thionyl chloride or catalytic hydrogenation setups simplifies the safety compliance burden, reducing the need for specialized equipment and extensive environmental remediation protocols. This streamlined workflow translates into a more resilient supply chain capable of responding quickly to market demands for anti-diabetic intermediates.
- Cost Reduction in Manufacturing: The primary economic driver of this technology is the drastic simplification of the synthetic route. By removing the necessity for amino protection groups and their subsequent removal, the process eliminates entire unit operations such as filtration, washing, and drying associated with intermediate isolations. This reduction in processing steps leads to a significant decrease in labor costs and facility occupancy time. Additionally, the high yields reported in the patent examples mean that less starting material is required to produce the same amount of final product, effectively lowering the cost per kilogram. The use of common, commodity-grade solvents like THF and ethyl acetate further ensures that operational expenses remain predictable and manageable, avoiding the volatility associated with exotic or highly regulated solvents.
- Enhanced Supply Chain Reliability: The reliance on readily available starting materials such as p-aminobenzoic acid and standard L-amino acids ensures a stable and secure supply chain. Unlike specialized protected building blocks that may have limited suppliers and long lead times, these commoditized inputs can be sourced from multiple vendors globally, mitigating the risk of supply disruptions. The robustness of the reaction conditions, which tolerate a broad temperature range and do not require strictly anhydrous or inert atmospheres beyond standard practice, makes the process highly transferable between different manufacturing sites. This flexibility allows for diversified production strategies, ensuring continuity of supply even in the face of regional logistical challenges or geopolitical instability affecting specific raw material sources.
- Scalability and Environmental Compliance: From an environmental and scalability perspective, this method aligns perfectly with green chemistry principles. The atom economy is improved by avoiding the mass-intensive protection-deprotection cycles, resulting in a lower E-factor (mass of waste per mass of product). The mild reaction temperatures reduce the energy load on heating and cooling systems, contributing to a smaller carbon footprint. Furthermore, the ease of purification, often achievable through simple recrystallization or standard column chromatography as noted in the patent, minimizes the volume of solvent waste generated during downstream processing. These factors collectively make the commercial scale-up of complex pharmaceutical intermediates more sustainable and compliant with increasingly stringent environmental regulations, positioning manufacturers as responsible partners in the global pharmaceutical value chain.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the synthesis and application of these pseudo-dipeptide esters. The answers are derived directly from the experimental data and technical disclosures within the patent documentation, providing reliable guidance for stakeholders evaluating this technology for their own development pipelines.
Q: Why is the direct coupling of unprotected p-aminobenzoic acid advantageous?
A: Traditional methods require tedious amino protection and deprotection steps or hazardous nitro reductions. This patent demonstrates that direct coupling omits these steps, significantly reducing operational complexity and improving overall yield.
Q: What coupling agents provide the best results for this synthesis?
A: Experimental data indicates that carbodiimides such as DIC or DCC, when used with HOBt as an additive, offer superior reaction rates, easier post-treatment, and higher yields compared to other activators like TBTU or EDC.
Q: Do these compounds exhibit biological activity?
A: Yes, screening results confirm that specific derivatives, particularly those with bulky side chains like the serine benzyl ester derivative, show potent PPAR agonistic activity, validating their potential as anti-diabetic drug candidates.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable P-Aminobenzoic Acid Pseudo-Dipeptide Ester Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of efficient and scalable synthesis routes in the race to bring new anti-diabetic therapies to market. Our team of expert process chemists has extensively evaluated the methodology described in CN101445468A and possesses the technical capability to optimize this direct coupling strategy for your specific needs. We boast extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your transition from lab bench to pilot plant and finally to full-scale manufacturing is seamless. Our state-of-the-art facilities are equipped with rigorous QC labs and advanced analytical instrumentation to guarantee stringent purity specifications, delivering intermediates that meet the highest global regulatory standards for pharmaceutical applications.
We invite you to collaborate with us to leverage this innovative synthesis technology for your next-generation drug development projects. Our technical procurement team is ready to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements and quality targets. Please contact us today to request specific COA data for our available inventory or to discuss route feasibility assessments for custom derivatives. By partnering with NINGBO INNO PHARMCHEM, you gain access to a reliable supply chain partner committed to driving down costs while maintaining the uncompromising quality essential for life-saving medications.