Advanced Synthesis of 3'-Azidothymidine Thiophosphoramidates for Antiviral Drug Development
The pharmaceutical landscape for antiviral therapeutics is constantly evolving, driven by the need for compounds that can withstand metabolic degradation while maintaining high potency. Patent CN1133641C introduces a significant advancement in this field by disclosing a series of thiophosphoryl amino acid ester compounds containing 3'-azidothymidine (AZT). Unlike traditional nucleoside analogs that suffer from rapid dephosphorylation, these novel prodrugs utilize a thiophosphoryl linkage to enhance stability against nucleases. The patent details a robust synthetic methodology that allows for the modular assembly of these complex molecules, offering a pathway to develop next-generation anti-AIDS and antitumor agents. For research and development teams, this technology represents a critical opportunity to access high-purity intermediates with proven biological activity in CEM and MT-4 cell lines.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the development of nucleoside reverse transcriptase inhibitors has been hampered by the inherent instability of phosphate esters in biological systems. Conventional phosphodiester linkages are readily recognized and cleaved by ubiquitous phosphatases and nucleases present in serum and within cells. This rapid enzymatic digestion often results in poor bioavailability, requiring frequent high-dose administration which can lead to increased toxicity and patient non-compliance. Furthermore, the synthesis of stable phosphate prodrugs often involves complex protecting group strategies that are difficult to scale and result in low overall yields. The inability to effectively deliver the active monophosphate species into the cytoplasm remains a primary bottleneck in the commercialization of many promising nucleoside candidates.
The Novel Approach
The methodology outlined in the patent overcomes these hurdles by employing a thiophosphoryl amino acid ester architecture. By replacing the bridging oxygen with sulfur, the resulting P-S bond exhibits remarkable resistance to enzymatic hydrolysis, effectively shielding the drug from premature degradation. The integration of an amino acid ester moiety further enhances the lipophilicity of the molecule, facilitating passive diffusion across cell membranes.  This structural innovation not only improves pharmacokinetic profiles but also simplifies the synthetic route. The process utilizes readily available starting materials such as aryl dichlorothiophosphates and amino acid methyl esters, reacting them under mild conditions to generate the target prodrugs efficiently. This approach provides a reliable pharmaceutical intermediate supplier with a versatile platform for generating diverse analogs tailored for specific therapeutic needs.
This structural innovation not only improves pharmacokinetic profiles but also simplifies the synthetic route. The process utilizes readily available starting materials such as aryl dichlorothiophosphates and amino acid methyl esters, reacting them under mild conditions to generate the target prodrugs efficiently. This approach provides a reliable pharmaceutical intermediate supplier with a versatile platform for generating diverse analogs tailored for specific therapeutic needs.
Mechanistic Insights into Thiophosphoryl Coupling Reactions
The core of this synthesis lies in the sequential nucleophilic substitution at the phosphorus center. The reaction initiates with the activation of the aryl dichlorothiophosphate in an anhydrous tetrahydrofuran (THF) environment. The first nucleophile, the amino acid methyl ester hydrochloride, attacks the phosphorus atom, displacing one chloride ion. This step is critically dependent on the presence of an acid-binding agent, typically triethylamine, which scavenges the generated hydrogen chloride to prevent the protonation of the amine nucleophile. The reaction is conducted at low temperatures, specifically between -4°C and -8°C, to kinetically control the formation of the mono-substituted intermediate and suppress the formation of bis-amino acid byproducts. This precise thermal management is essential for maintaining the integrity of the sensitive thiophosphoryl chloride functionality.
Following the formation of the amino acid-thiophosphoryl intermediate, the second substitution occurs with the introduction of 3'-azidothymidine. The 5'-hydroxyl group of the AZT acts as the second nucleophile, attacking the phosphorus center to displace the remaining chloride. 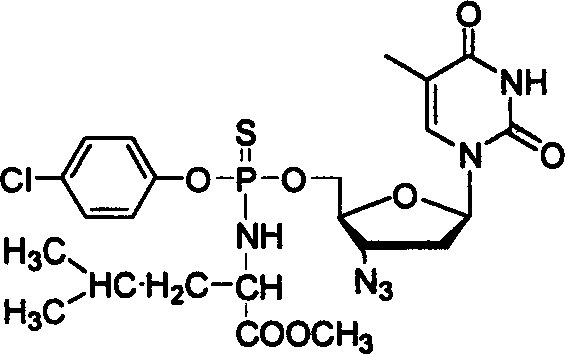 The use of NMR spectroscopy to monitor the reaction progress ensures that the first step is complete before the addition of the valuable nucleoside, maximizing atom economy and minimizing waste. The final product is isolated via silica gel column chromatography, yielding compounds with distinct spectral signatures, such as characteristic 31P NMR shifts around 70-72 ppm. This rigorous control over the reaction mechanism ensures consistent impurity profiles, which is vital for cost reduction in API manufacturing where downstream purification costs can be prohibitive.
The use of NMR spectroscopy to monitor the reaction progress ensures that the first step is complete before the addition of the valuable nucleoside, maximizing atom economy and minimizing waste. The final product is isolated via silica gel column chromatography, yielding compounds with distinct spectral signatures, such as characteristic 31P NMR shifts around 70-72 ppm. This rigorous control over the reaction mechanism ensures consistent impurity profiles, which is vital for cost reduction in API manufacturing where downstream purification costs can be prohibitive.
How to Synthesize 3'-Azidothymidine Thiophosphoramidates Efficiently
The synthesis protocol described in the patent offers a streamlined route for producing these high-value intermediates. The process begins with the preparation of a 0.8 to 1.0 mol/L solution of the aryl substituted dichlorothiophosphate in dried THF under an inert nitrogen atmosphere. Strict adherence to the temperature range of -4°C to -8°C is maintained throughout the addition of reagents. The amino acid component is added first, followed by the base, and finally the AZT solution. This ordered addition is crucial for directing the regioselectivity of the phosphorylation.
- Dissolve the aryl substituted dichlorothiophosphate in dried tetrahydrofuran (THF) under nitrogen protection and cool the solution to between -4°C and -8°C using an ice-salt bath.
- Add the amino acid methyl ester hydrochloride to the cooled solution, followed by the dropwise addition of an acid-binding agent such as triethylamine to neutralize the generated HCl.
- Once the initial reaction is complete, slowly add the 3'-azidothymidine (AZT) solution dissolved in THF, followed by a second equivalent of the acid-binding agent to drive the coupling to completion.
- Monitor the reaction via NMR, then filter, remove solvents by rotary distillation, and purify the crude product using silica gel column chromatography with a chloroform-methanol eluent.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this synthetic route offers substantial strategic benefits beyond mere technical feasibility. The reliance on commodity chemicals such as THF, triethylamine, and standard amino acid esters significantly mitigates supply chain risks associated with exotic or proprietary reagents. The process avoids the use of expensive transition metal catalysts or enzymatic steps that often complicate scale-up and regulatory approval. Instead, it utilizes standard unit operations like filtration, rotary distillation, and column chromatography, which are well-understood and easily implemented in existing GMP facilities. This compatibility with standard infrastructure translates directly into reduced capital expenditure and faster time-to-market for new drug candidates.
- Cost Reduction in Manufacturing: The synthetic pathway eliminates the need for complex protecting group manipulations that are typical in traditional oligonucleotide synthesis. By utilizing a direct coupling strategy in a single pot for each substitution step, the number of isolation and purification stages is minimized. This reduction in processing steps leads to significant savings in solvent consumption, labor hours, and waste disposal costs. Furthermore, the high yields reported in the patent examples, ranging from approximately 60% to over 80% depending on the specific amino acid variant, ensure efficient utilization of the expensive nucleoside starting material, thereby optimizing the overall cost of goods sold.
- Enhanced Supply Chain Reliability: The starting materials for this synthesis, including various aryl dichlorothiophosphates and amino acid methyl esters, are commercially available in bulk quantities from multiple global suppliers. This diversity in sourcing options prevents bottlenecks and ensures continuity of supply even during market fluctuations. The robustness of the reaction conditions, particularly the tolerance for standard laboratory temperatures once the initial cooling phase is managed, allows for flexible production scheduling. This reliability is critical for maintaining the inventory levels required for clinical trial material production and eventual commercial launch.
- Scalability and Environmental Compliance: The process is inherently scalable, moving seamlessly from gram-scale laboratory experiments to multi-kilogram pilot runs. The use of THF as a primary solvent is advantageous as it is easily recovered and recycled through distillation, aligning with green chemistry principles and reducing the environmental footprint of the manufacturing process. The absence of heavy metal contaminants simplifies the purification workflow and reduces the burden on quality control laboratories to perform extensive residual metal testing. This streamlined compliance profile accelerates the regulatory filing process and supports sustainable manufacturing practices.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of these thiophosphoryl compounds. Understanding these details is essential for partners looking to integrate this technology into their development pipelines.
Q: Why are thiophosphoryl amino acid esters preferred over natural phosphodiesters for AZT prodrugs?
A: Natural phosphodiester bonds in oligonucleotides are highly susceptible to degradation by intracellular and extracellular nucleases, which rapidly digest the drug before it can act. The thiophosphoryl modification replaces a non-bridging oxygen with sulfur, creating a P-S bond that confers significant resistance to nuclease digestion, thereby enhancing the stability and biological half-life of the antiviral agent.
Q: What is the critical temperature range for the synthesis described in Patent CN1133641C?
A: The synthesis requires strict temperature control, specifically cooling the reaction mixture to between -4°C and -8°C using an ice-salt bath. This low-temperature environment is essential to control the reactivity of the dichlorothiophosphate intermediate, preventing side reactions such as hydrolysis or over-substitution, and ensuring high purity of the final thiophosphoramidate product.
Q: How does the amino acid moiety contribute to the efficacy of these compounds?
A: The amino acid ester serves as a lipophilic promoiety that enhances the cell membrane permeability of the charged phosphate analog. Once inside the cell, enzymatic hydrolysis releases the active monophosphate form of the nucleoside. Different amino acids (glycine, alanine, phenylalanine, etc.) allow for tuning the lipophilicity and metabolic stability of the prodrug.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 3'-Azidothymidine Thiophosphoramidate Supplier
At NINGBO INNO PHARMCHEM, we recognize the transformative potential of the chemistry described in Patent CN1133641C for the development of advanced antiviral therapies. As a leading CDMO partner, we possess the technical expertise and infrastructure to translate these laboratory protocols into commercial reality. Our team has extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your supply needs are met with precision and consistency. We operate stringent purity specifications and maintain rigorous QC labs equipped with state-of-the-art analytical instrumentation to guarantee the quality of every batch of 3'-azidothymidine thiophosphoramidate we produce.
We invite you to collaborate with us to optimize this synthesis for your specific project requirements. Our technical procurement team is ready to provide a Customized Cost-Saving Analysis that evaluates the economic viability of this route compared to your current methods. Contact us today to request specific COA data and route feasibility assessments, and let us help you accelerate the development of your next-generation antiviral drug candidate.