Advanced Manufacturing of High-Purity Leucogen: Overcoming Impurity Challenges in Pharmaceutical Intermediates
Advanced Manufacturing of High-Purity Leucogen: Overcoming Impurity Challenges in Pharmaceutical Intermediates
The pharmaceutical landscape demands increasingly rigorous standards for active pharmaceutical ingredients (APIs) and their precursors, particularly in the treatment of hematological disorders where purity is paramount. Patent CN103102324A introduces a groundbreaking preparation method for Leucogen, a critical therapeutic agent used to manage leukopenia and other blood system diseases. This innovation addresses long-standing challenges in the synthesis of Leucogen by fundamentally re-engineering the condensation and purification stages to eliminate specific genotoxic impurities. By shifting away from traditional vacuum distillation techniques towards a sophisticated extraction and crystallization protocol, this technology ensures the final product is free from methyl ester impurities that have historically plagued manufacturing batches. For R&D directors and procurement specialists seeking a reliable pharmaceutical intermediates supplier, understanding the mechanistic superiority of this route is essential for securing a stable, high-quality supply chain.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
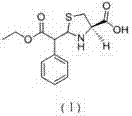
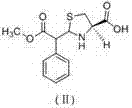
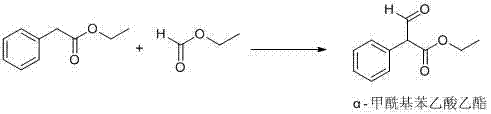
Historically, the synthesis of the key intermediate, ethyl alpha-formylphenylacetate, relied heavily on prolonged reaction times and energy-intensive purification strategies that introduced significant quality risks. Prior art, such as that disclosed in CN1872844A and CN1810791A, typically required reaction durations extending up to 10 hours, creating a bottleneck in production throughput and increasing operational costs. More critically, these conventional processes depended on vacuum distillation to remove reaction solvents, a step that often failed to completely eliminate residual ethyl phenylacetate. This residual starting material would subsequently react with L-cysteine hydrochloride in the second stage, leading to the formation of a persistent methyl ester impurity, structurally identified as compound (II). The presence of this impurity, often exceeding 0.1%, rendered the final Leucogen bulk drug non-compliant with stringent medicinal purity requirements, necessitating costly re-processing or batch rejection.
The Novel Approach
The innovative methodology presented in CN103102324A circumvents these pitfalls by optimizing both the reaction kinetics and the downstream processing workflow. By employing specific ether solvents such as isopropyl ether or methyl tert-butyl ether (MTBE) in conjunction with strong alkali metal bases like sodium tert-butoxide, the condensation reaction is accelerated significantly, completing within a concise 4 to 6-hour window. Instead of relying on thermal separation via distillation, the new protocol utilizes a liquid-liquid extraction followed by controlled acidification and low-temperature crystallization. This physical purification strategy effectively removes unreacted starting materials and by-products before they can interfere with the subsequent cyclization step. The result is a high-purity solid intermediate that reacts cleanly with L-cysteine, ensuring the final Leucogen product meets all pharmacopeial standards without the detectable presence of the problematic methyl ester impurity.
Mechanistic Insights into Tert-Butoxide Catalyzed Condensation
The core of this technological advancement lies in the precise selection of the condensing agent and solvent system, which fundamentally alters the reaction pathway and endpoint visibility. In this optimized Claisen-type condensation, the use of bulky tert-butoxide bases in aprotic ether solvents creates a highly reactive enolate environment that facilitates rapid nucleophilic attack on the formyl carbon of ethyl formate. This specific chemical environment not only accelerates the reaction rate but also induces a distinct physical change in the reaction mixture; the transition to a white opaque suspension serves as a robust, visual indicator of reaction completion. This phenomenon eliminates the dependency on resource-intensive analytical monitoring techniques such as Thin Layer Chromatography (TLC) or High-Performance Liquid Chromatography (HPLC) for endpoint determination, thereby streamlining the operational workflow for plant technicians and reducing the potential for human error during batch termination.
Furthermore, the purification mechanism plays a pivotal role in controlling the impurity profile of the final API intermediate. By adjusting the pH of the aqueous phase to between 1.0 and 2.0 using dilute hydrochloric acid, the process ensures the selective protonation and precipitation of the desired alpha-formyl phenylacetic acid ethyl ester while leaving polar impurities in the solution. Subsequent crystallization at temperatures between 0°C and 5°C further refines the crystal lattice, excluding structurally similar contaminants that might otherwise co-precipitate at higher temperatures. This rigorous control over the solid-state properties of the intermediate is crucial because it prevents the carryover of ethyl phenylacetate into the second reaction vessel. Consequently, when the intermediate reacts with L-cysteine hydrochloride in a moist acetone system, the pathway to forming the methyl ester side-product is chemically blocked, guaranteeing a final product with a total impurity profile of less than 0.3% and a main isomer content exceeding 95%.
How to Synthesize Ethyl Alpha-Formylphenylacetate Efficiently
The synthesis of the critical intermediate, ethyl alpha-formylphenylacetate, serves as the foundation for producing high-quality Leucogen and requires strict adherence to the optimized parameters defined in the patent. The process begins with the preparation of a basic ether solution, followed by the controlled addition of esters under cooling to manage the exotherm, and concludes with a careful workup to isolate the solid product. While the general concept of condensation is well-known, the specific combination of solvents, bases, and temperature ramps described here is what enables the commercial viability of the route. For detailed laboratory protocols and exact stoichiometric ratios required to replicate this high-yield process, please refer to the standardized synthesis steps outlined below.
- Perform condensation of ethyl phenylacetate and ethyl formate in isopropyl ether or MTBE using sodium or potassium tert-butoxide at 5-30°C.
- Purify the intermediate alpha-formyl phenylacetic acid ethyl ester via water-ether extraction, acidification to pH 1.0-2.0, and low-temperature crystallization.
- React the purified intermediate with L-cysteine hydrochloride in a moist acetone system under reflux to obtain Leucogen.
Commercial Advantages for Procurement and Supply Chain Teams
From a strategic sourcing perspective, the adoption of this patented synthesis route offers substantial benefits that extend far beyond simple chemical yield improvements. For procurement managers and supply chain heads, the elimination of complex unit operations translates directly into reduced capital expenditure and lower operating costs, making the final API intermediate more price-competitive in the global market. The simplified workflow reduces the dependency on specialized vacuum distillation equipment, which in turn lowers maintenance overheads and minimizes the risk of production downtime due to equipment failure. Furthermore, the robustness of the crystallization-based purification ensures consistent batch-to-batch quality, reducing the likelihood of supply disruptions caused by out-of-specification results and enhancing the overall reliability of the supply chain for downstream drug manufacturers.
- Cost Reduction in Manufacturing: The transition from vacuum distillation to extraction and crystallization represents a significant decrease in energy consumption, as thermal separation processes are notoriously energy-intensive compared to ambient or low-temperature physical separations. By removing the need for prolonged heating and high-vacuum conditions, the facility can achieve drastic reductions in utility costs, including steam and electricity usage, which are major components of the cost of goods sold (COGS). Additionally, the shorter reaction time of 4 to 6 hours increases the turnover rate of the reactor vessels, allowing for more batches to be produced within the same timeframe without requiring additional capital investment in new hardware. This intensification of the process leads to substantial cost savings that can be passed down to the customer or reinvested into further quality assurance measures.
- Enhanced Supply Chain Reliability: The visual endpoint indicator provided by the formation of a white opaque suspension simplifies process control and reduces the reliance on external laboratory support for real-time decision-making. This operational simplicity minimizes the risk of batch failures due to over-reaction or under-reaction, ensuring a more predictable and steady output of the intermediate. Moreover, the use of common, commercially available solvents like isopropyl ether and MTBE mitigates the risk of raw material shortages that can occur with exotic or highly regulated reagents. This accessibility of inputs ensures that production schedules can be maintained even during periods of market volatility, providing downstream partners with a secure and continuous supply of critical pharmaceutical materials.
- Scalability and Environmental Compliance: The avoidance of high-temperature distillation steps inherently reduces the generation of volatile organic compound (VOC) emissions, aligning the manufacturing process with increasingly stringent environmental regulations and sustainability goals. The wastewater generated from the extraction and acidification steps is easier to treat compared to the complex residues often associated with thermal degradation products from distillation bottoms. This environmental friendliness facilitates smoother regulatory approvals and reduces the liability associated with waste disposal. Furthermore, the crystallization step is inherently scalable; the physics of crystal growth remain consistent from the kilogram to the tonne scale, ensuring that the high purity achieved in the lab can be reliably reproduced in commercial-scale reactors ranging from 100 kgs to 100 MT annual production capacities.
Frequently Asked Questions (FAQ)
To assist technical teams in evaluating the feasibility of integrating this technology into their existing supply chains, we have compiled answers to common inquiries regarding the process specifications and quality outcomes. These responses are derived directly from the experimental data and technical disclosures within the patent documentation, providing a transparent view of the method's capabilities. Understanding these details is crucial for assessing the compatibility of this intermediate with your specific formulation requirements and regulatory filings.
Q: How does the new method prevent methyl ester impurity formation in Leucogen?
A: The novel process avoids vacuum distillation of the reaction solvent, which traditionally left residual ethyl phenylacetate. By using extraction and low-temperature crystallization, the intermediate purity is significantly enhanced, preventing the formation of methyl ester impurity (II) during the subsequent cysteine reaction.
Q: What are the advantages of using tert-butoxide bases in ether solvents?
A: Using sodium or potassium tert-butoxide in isopropyl ether or MTBE shortens reaction time to 4-6 hours compared to 10 hours in conventional methods. It also provides a clear visual endpoint (white opacity), simplifying process monitoring without needing extensive TLC or HPLC analysis.
Q: Is this Leucogen synthesis method suitable for industrial scale-up?
A: Yes, the method is designed for industrial suitability. It eliminates complex vacuum distillation steps, uses common ether solvents, and relies on simple crystallization for purification, making it robust for large-scale commercial production of pharmaceutical intermediates.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Leucogen Supplier
At NINGBO INNO PHARMCHEM, we recognize that the transition from patent literature to commercial reality requires a partner with deep technical expertise and robust manufacturing infrastructure. As a leading CDMO, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the theoretical benefits of the CN103102324A process are fully realized in a GMP-compliant environment. Our facilities are equipped with state-of-the-art reaction vessels capable of handling the specific solvent systems and low-temperature crystallization requirements of this synthesis, backed by rigorous QC labs that enforce stringent purity specifications to guarantee every batch meets the highest international standards.
We invite pharmaceutical companies and research institutions to collaborate with us to leverage this advanced synthesis route for their hematology drug portfolios. By partnering with our technical procurement team, you can request a Customized Cost-Saving Analysis that quantifies the economic benefits of switching to this impurity-free process. We encourage you to contact us today to obtain specific COA data from our pilot batches and to discuss route feasibility assessments tailored to your project timelines, ensuring a seamless path from development to market launch.