Advanced Silane Reduction Technology for Commercial Scale Dibenzylbiotin Manufacturing
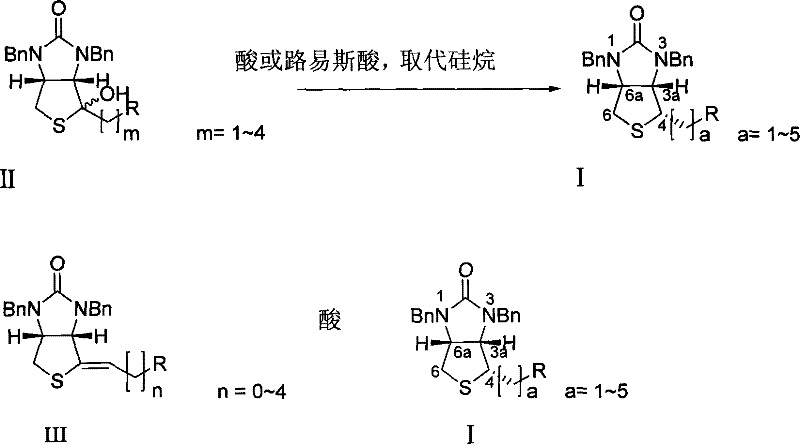
The pharmaceutical industry constantly seeks robust and scalable pathways for essential vitamin intermediates, and patent CN101215292B presents a transformative approach to the synthesis of dibenzylbiotin and its derivatives. This specific intellectual property outlines a highly efficient chemical transformation that converts tertiary alcohol or alkene precursors into the target biotin intermediate using substituted silanes under acidic conditions. Unlike traditional methods that rely on harsh catalytic hydrogenation, this novel protocol operates under remarkably mild conditions, typically ranging from cryogenic temperatures up to moderate warmth, ensuring the integrity of sensitive functional groups. The technology represents a significant leap forward in process chemistry, offering a route that is not only chemically elegant but also industrially viable for the production of high-purity Vitamin H precursors. By leveraging the unique reactivity of silanes in the presence of Lewis or Brønsted acids, manufacturers can achieve exceptional yields exceeding 90 percent while maintaining strict stereochemical control. This development is particularly critical for global supply chains aiming to secure reliable sources of complex heterocyclic intermediates without the bottlenecks associated with noble metal dependency.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the construction of the third chiral center in biotin synthesis has been dominated by the classical Goldberg and Sternbach methodology, which relies heavily on catalytic hydrogenation steps. While effective in laboratory settings, this traditional approach suffers from severe industrial drawbacks, primarily due to the susceptibility of palladium catalysts to poisoning by the sulfur atom inherent in the biotin heterocyclic core. This sulfur poisoning effect drastically reduces catalyst life and efficiency, necessitating frequent replacement of expensive noble metals and leading to inconsistent batch-to-batch performance. Furthermore, conventional hydrogenation requires specialized high-pressure reactor vessels and elevated temperatures to drive the reaction to completion, introducing significant safety hazards and capital expenditure requirements for manufacturing facilities. The need for rigorous removal of trace palladium residues from the final active pharmaceutical ingredient adds further complexity and cost to the downstream purification process. These cumulative factors create a fragile supply chain where production delays and quality deviations are common, making the traditional route suboptimal for modern large-scale commercial manufacturing demands.
The Novel Approach
In stark contrast, the methodology disclosed in CN101215292B circumvents these historical challenges by employing a biomimetic-style reduction using substituted silanes as the hydride source. This innovative strategy completely eliminates the requirement for transition metal catalysts, thereby removing the risk of sulfur poisoning and the associated need for costly metal scavenging steps. The reaction proceeds smoothly in common organic solvents such as dichloromethane or tetrahydrofuran under atmospheric pressure, utilizing readily available acids like trifluoroacetic acid or boron trifluoride etherate to activate the substrate. This shift from heterogeneous catalysis to homogeneous chemical reduction allows for much finer control over reaction parameters, resulting in superior impurity profiles and higher overall throughput. The mildness of the conditions ensures that sensitive ester or acid side chains on the valeric acid moiety remain intact, preventing unwanted side reactions that often plague high-energy hydrogenation processes. Consequently, this novel approach offers a streamlined, safer, and more economically attractive pathway for the industrial production of dibenzylbiotin derivatives.
Mechanistic Insights into Lewis Acid Catalyzed Silane Reduction
The core of this synthetic breakthrough lies in the precise activation of the substrate by the acid catalyst, which facilitates a highly stereoselective hydride transfer from the silane reagent. In the case of the tertiary alcohol precursor, the Lewis acid or protonic acid coordinates with the hydroxyl group, promoting the formation of a stabilized carbocation intermediate or an ion-pair species at the C4 position. This activation step is crucial as it renders the carbon center electrophilic enough to accept a hydride ion from the silicon-hydrogen bond of the triethylsilane or similar reagent. The geometry of the cis-fused thieno[3,4-d]imidazolidinone ring system plays a pivotal role in directing the incoming hydride, ensuring that the reduction occurs exclusively from the alpha-face to establish the desired 4S configuration. This inherent substrate control minimizes the formation of diastereomeric impurities, which are notoriously difficult to separate in later stages of synthesis. For the alkene precursor variant, the mechanism likely involves protonation of the double bond to generate a similar cationic intermediate, which is then trapped by the silane hydride in a concerted or stepwise fashion. The versatility of this mechanism allows it to accommodate various substituents on the side chain, demonstrating broad scope and robustness across different derivative structures.
From an impurity control perspective, this mechanism offers distinct advantages over radical-based or metal-catalyzed reductions which often lead to over-reduction or ring-opening byproducts. The electrophilic nature of the activation ensures that the reaction is chemoselective for the specific tertiary center or alkene, leaving other reducible functionalities such as esters or amides untouched. This high level of chemoselectivity simplifies the workup procedure, often requiring only a basic wash to neutralize the acid and remove silicon byproducts, followed by solvent evaporation. The absence of metal particulates means that filtration steps are straightforward, and the risk of metal contamination in the final API is virtually non-existent. Furthermore, the reaction kinetics are favorable, typically reaching completion within 8 to 24 hours at temperatures that do not require extreme cooling or heating, which enhances energy efficiency. The ability to tune the acidity and the steric bulk of the silane provides process chemists with additional handles to optimize the reaction for specific scale-up requirements, ensuring consistent quality regardless of batch size.
How to Synthesize Dibenzylbiotin Efficiently
The practical execution of this synthesis involves dissolving the starting material, either the tertiary alcohol or the alkene derivative, in a dry aprotic solvent such as dichloromethane under an inert atmosphere. The reaction mixture is then cooled to a temperature between -70°C and 0°C to manage the exotherm upon addition of the acid catalyst, which is typically added dropwise to ensure uniform mixing and prevent localized hot spots. Following the activation period, the substituted silane is introduced, and the reaction is allowed to warm slowly to room temperature over a period of several hours to drive the conversion to completion. Detailed standardized operating procedures regarding specific molar ratios, quenching protocols, and isolation techniques are critical for reproducibility and are outlined in the comprehensive guide below.
- Prepare the reaction vessel with the tertiary alcohol or alkene precursor and an appropriate organic solvent such as dichloromethane or tetrahydrofuran.
- Cool the reaction mixture to temperatures between -80°C and 0°C, then add the Lewis acid or protonic acid catalyst followed by the substituted silane reducing agent.
- Allow the reaction to proceed while warming to room temperature over several hours, then quench with aqueous base and isolate the product via standard extraction and purification techniques.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this silane-based reduction technology translates directly into enhanced operational stability and reduced total cost of ownership. By removing the dependency on palladium catalysts, manufacturers eliminate a major variable cost component that is subject to volatile market pricing and geopolitical supply risks associated with precious metals. The simplified equipment requirements mean that production can be scheduled on standard glass-lined or stainless steel reactors without the need for specialized high-pressure autoclaves, thereby increasing facility utilization rates and flexibility. This process intensification allows for faster turnaround times between batches, significantly reducing the lead time for high-purity pharmaceutical intermediates needed for downstream formulation. Additionally, the high yield and selectivity reported in the patent minimize raw material waste, contributing to a more sustainable and cost-effective manufacturing footprint that aligns with modern green chemistry initiatives.
- Cost Reduction in Manufacturing: The elimination of expensive palladium catalysts and the associated metal scavenging resins results in substantial direct material savings for every kilogram of product produced. Furthermore, the avoidance of high-pressure hydrogenation equipment reduces both capital depreciation costs and maintenance expenses related to safety inspections and reactor certifications. The use of commodity chemicals like triethylsilane and trifluoroacetic acid ensures that raw material costs remain stable and predictable, shielding the supply chain from the price fluctuations typical of noble metals. Process efficiency is further enhanced by the high conversion rates, which reduce the volume of solvent and energy required per unit of output, driving down utility costs significantly.
- Enhanced Supply Chain Reliability: Sourcing silanes and common organic acids is far less complex than securing specialized hydrogenation catalysts, which often have long lead times and limited supplier bases. This diversification of the supply base mitigates the risk of production stoppages due to raw material shortages, ensuring a continuous flow of intermediates to downstream customers. The robustness of the reaction conditions means that production is less susceptible to minor variations in utility supplies or environmental conditions, leading to more reliable delivery schedules. Consequently, partners can maintain lower safety stock levels while still meeting just-in-time delivery commitments, optimizing working capital and inventory management strategies across the value chain.
- Scalability and Environmental Compliance: The mild reaction conditions and lack of heavy metal waste streams simplify the environmental permitting process and reduce the burden on wastewater treatment facilities. Scaling this process from pilot plant to commercial tonnage is straightforward since it does not involve the complex heat and mass transfer limitations often encountered in gas-liquid hydrogenation reactions. The reduced generation of hazardous waste aligns with increasingly stringent global environmental regulations, minimizing the risk of compliance penalties and enhancing the corporate sustainability profile. This ease of scale-up ensures that supply can be rapidly ramped up to meet surging market demand for biotin-containing nutraceuticals and pharmaceuticals without compromising on quality or safety standards.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this novel synthesis route. These answers are derived directly from the experimental data and claims presented in the patent documentation to provide clarity on process capabilities and limitations. Understanding these details is essential for technical teams evaluating the feasibility of integrating this technology into their existing manufacturing portfolios.
Q: What are the primary advantages of the silane reduction method over traditional hydrogenation for biotin intermediates?
A: The silane reduction method described in CN101215292B eliminates the need for expensive palladium catalysts which are prone to sulfur poisoning. It also operates under mild atmospheric pressure conditions rather than requiring high-pressure hydrogenation equipment, significantly improving safety and reducing capital expenditure.
Q: What is the stereochemical outcome of this reduction process?
A: The process utilizes the inherent stereocontrol of the cis-fused thieno-imidazolidinone ring system to achieve high stereoselectivity. The hydride attack occurs from the less hindered face directed by the existing chiral centers at positions 3a and 6a, yielding the desired single configuration product with high optical purity.
Q: Which reagents are critical for the success of this synthesis?
A: The key reagents include substituted silanes such as triethylsilane or trimethylsilane acting as the hydride source, and Lewis acids like boron trifluoride etherate or protonic acids like trifluoroacetic acid to activate the substrate. Common solvents like dichloromethane or THF are used to facilitate the reaction.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Dibenzylbiotin Supplier
At NINGBO INNO PHARMCHEM, we recognize the strategic importance of efficient intermediate synthesis in the competitive landscape of vitamin and pharmaceutical manufacturing. Our technical team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the benefits of advanced patents like CN101215292B are fully realized at an industrial level. We operate stringent purity specifications and maintain rigorous QC labs equipped with state-of-the-art analytical instrumentation to guarantee that every batch of dibenzylbiotin meets the highest international standards. Our commitment to process excellence allows us to deliver materials that facilitate smoother downstream processing for our clients, ultimately accelerating their time to market for finished dosage forms.
We invite potential partners to engage with our technical procurement team to discuss how this optimized synthesis route can benefit your specific project requirements. By requesting a Customized Cost-Saving Analysis, you can gain deeper insights into the economic advantages of switching to this metal-free reduction technology. We encourage you to contact us today to obtain specific COA data and route feasibility assessments tailored to your volume needs, ensuring a seamless integration of our high-quality intermediates into your supply chain.