Advanced Palladium-Catalyzed Synthesis of 3-Benzylidene-2,3-dihydroquinolone Intermediates for Scalable Pharma Manufacturing
Introduction to Next-Generation Quinolone Synthesis
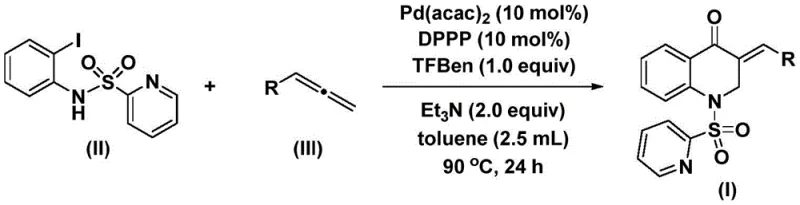
The development of efficient synthetic routes for nitrogen-containing heterocycles remains a cornerstone of modern medicinal chemistry, particularly for scaffolds exhibiting potent biological activities. As detailed in the recent patent publication CN113735826A, a groundbreaking preparation method for 3-benzylidene-2,3-dihydroquinolone compounds has been established, addressing critical bottlenecks in traditional synthesis. These quinolone derivatives serve as vital precursors for a wide array of bioactive molecules, including analgesic agents and antitumor drugs, making their reliable production essential for the global pharmaceutical supply chain. The disclosed technology leverages a sophisticated palladium-catalyzed carbonylation strategy that transforms readily available N-pyridinesulfonyl-o-iodoaniline and allene substrates into complex heterocyclic frameworks with remarkable precision.
This innovative approach not only streamlines the construction of the quinolone core but also introduces a level of operational simplicity that is rarely seen in transition metal-catalyzed carbonylations. By utilizing a solid carbon monoxide substitute instead of hazardous gas, the process significantly enhances laboratory and plant safety profiles. For R&D directors and process chemists, this represents a pivotal shift towards safer, more scalable methodologies that can be seamlessly integrated into existing manufacturing workflows. The versatility of this reaction allows for the rapid generation of diverse compound libraries, accelerating the drug discovery timeline for new therapeutic candidates targeting pain management and oncology.
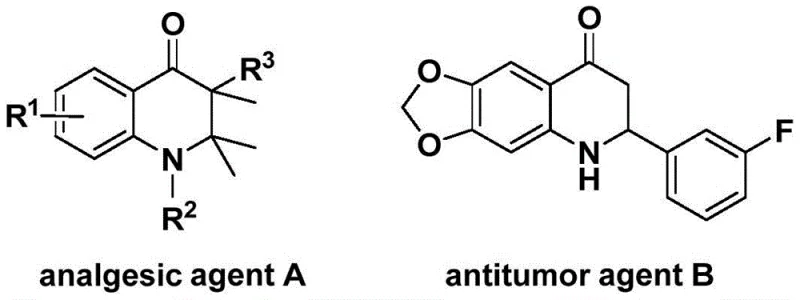
The structural diversity achievable through this method is exemplified by the core scaffold's presence in known pharmacophores. As illustrated in the reference structures, the 2,3-dihydroquinolone motif is a privileged structure found in potent analgesic agent A and antitumor agent B. The ability to functionalize the 3-position with various benzylidene groups opens up vast chemical space for structure-activity relationship (SAR) studies. This patent provides the key to unlocking these structures efficiently, offering a robust platform for the synthesis of high-value pharmaceutical intermediates that were previously difficult or expensive to access through conventional cyclization protocols.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of 2,3-dihydroquinolone compounds has relied on classical condensation reactions or multi-step sequences that often suffer from poor atom economy and harsh reaction conditions. Traditional methods frequently require strong acids or bases, elevated temperatures exceeding 150°C, and prolonged reaction times that can lead to the degradation of sensitive functional groups. Furthermore, many existing protocols struggle with regioselectivity issues, resulting in complex mixtures of isomers that are challenging and costly to separate on an industrial scale. The reliance on gaseous carbon monoxide in some carbonylation approaches introduces significant safety hazards and engineering complexities, necessitating specialized high-pressure equipment that limits accessibility for many contract research organizations.
Another critical drawback of prior art is the limited substrate scope, particularly regarding the introduction of substituents at the 3-position. Conventional routes often fail to accommodate sterically hindered or electronically diverse allene-like precursors, restricting the chemical diversity available to medicinal chemists. This limitation forces R&D teams to settle for suboptimal analogs or invest excessive resources in developing custom synthetic routes for each new target. Additionally, the purification of products from these older methods often involves tedious workups and extensive chromatography, leading to substantial material loss and increased waste generation, which contradicts the principles of green chemistry increasingly demanded by regulatory bodies and corporate sustainability goals.
The Novel Approach
In stark contrast, the novel palladium-catalyzed protocol described in the patent data offers a transformative solution by employing a mild, one-pot tandem reaction sequence. This method utilizes N-pyridinesulfonyl-o-iodoaniline as a bifunctional precursor that undergoes oxidative addition with the palladium catalyst, followed by CO insertion and subsequent reaction with an allene. The use of 1,3,5-trimesic acid phenol ester (TFBen) as a solid CO surrogate eliminates the need for handling toxic carbon monoxide gas, thereby drastically reducing safety risks and infrastructure costs. The reaction proceeds smoothly in toluene at a moderate temperature of 90°C, conditions that are compatible with a broad range of functional groups including halogens, ethers, and alkyl chains.
The efficiency of this new approach is underscored by its exceptional yields and short reaction times relative to the complexity of the transformation. Experimental data indicates that the reaction reaches completion within 24 to 48 hours, delivering products with yields often exceeding 80% and reaching as high as 93% for optimized substrates. The catalytic system, comprising Pd(acac)2 and the bidentate ligand DPPP, ensures high turnover numbers and minimizes the residual metal content in the final product, a critical parameter for pharmaceutical grade materials. This streamlined process not only reduces the number of unit operations but also simplifies the downstream processing, allowing for direct filtration and standard column chromatography to achieve high purity levels suitable for further biological evaluation or scale-up.
Mechanistic Insights into Palladium-Catalyzed Carbonylative Cyclization
The success of this synthesis hinges on a meticulously orchestrated catalytic cycle that maximizes the reactivity of the palladium center while minimizing off-cycle decomposition pathways. The mechanism initiates with the oxidative addition of the palladium(0) species into the carbon-iodine bond of the N-pyridinesulfonyl-o-iodoaniline substrate. This step generates a reactive aryl-palladium(II) intermediate, which is stabilized by the coordination of the 1,3-bis(diphenylphosphino)propane (DPPP) ligand. The choice of DPPP is crucial, as its bite angle and electronic properties facilitate the subsequent migratory insertion of carbon monoxide. The CO molecule, released in situ from the thermal decomposition of TFBen, inserts into the Pd-C bond to form an acyl-palladium intermediate, effectively building the carbonyl functionality of the quinolone ring.
Following the carbonylation step, the allene substrate coordinates to the palladium center and undergoes migratory insertion into the acyl-palladium bond. This step is stereoselective and regioselective, driven by the steric and electronic environment created by the ligand and the sulfonyl group on the nitrogen. The resulting alkyl-palladium intermediate then undergoes an intramolecular nucleophilic attack or reductive elimination sequence to close the six-membered ring, releasing the final 3-benzylidene-2,3-dihydroquinolone product and regenerating the active palladium(0) catalyst. This elegant cascade avoids the formation of stable off-cycle species, ensuring high catalytic efficiency. The presence of triethylamine as a base likely assists in neutralizing acidic byproducts and maintaining the catalytic cycle's integrity throughout the 24-hour reaction period.
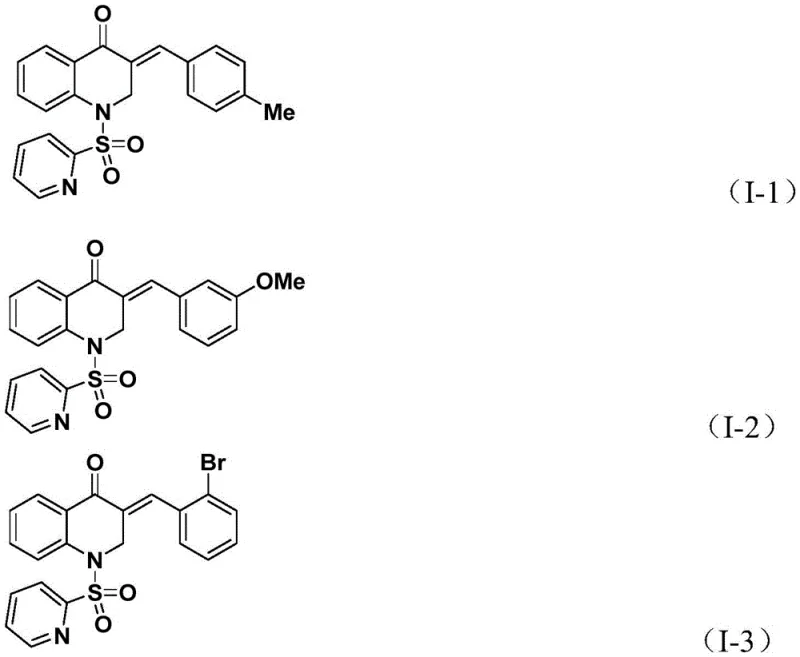
Impurity control is inherently built into this mechanism through the high specificity of the palladium insertion steps. Unlike radical-based cyclizations that can lead to polymerization or non-specific coupling, this organometallic pathway directs the reaction exclusively towards the desired heterocyclic product. The patent data confirms that even with diverse substituents on the allene moiety—ranging from electron-rich 4-methoxyphenyl groups to electron-deficient 4-chlorophenyl and 3-bromophenyl groups—the reaction maintains high fidelity. For instance, substrates with ortho-substitution or bulky naphthyl groups also proceed efficiently, yielding products like I-5 in 93% yield. This robustness suggests that the transition state for the allene insertion is sufficiently flexible to accommodate steric bulk without compromising the reaction rate or selectivity, a feature that is highly valuable for synthesizing complex drug candidates.
How to Synthesize 3-Benzylidene-2,3-dihydroquinolone Efficiently
Implementing this synthesis in a laboratory or pilot plant setting requires careful attention to reagent quality and reaction parameters to replicate the high yields reported in the patent. The process is designed to be operationally simple, requiring standard Schlenk techniques or sealed vessel reactors capable of maintaining 90°C. The key to success lies in the precise stoichiometry of the catalyst system and the use of dry, degassed toluene to prevent catalyst deactivation. Operators should ensure that the TFBen is fresh and that the palladium precursor is fully dissolved before heating to guarantee uniform initiation of the catalytic cycle. Detailed standardized synthetic steps for this procedure are provided in the guide below to assist technical teams in rapid adoption.
- Combine palladium catalyst (Pd(acac)2), DPPP ligand, TFBen (CO source), triethylamine, N-pyridinesulfonyl-o-iodoaniline, and allene substrate in toluene solvent within a Schlenk tube.
- Heat the reaction mixture to 90°C and maintain stirring for 24 to 48 hours to ensure complete conversion of the starting materials into the target quinolone scaffold.
- Upon completion, filter the mixture, mix with silica gel, and purify via column chromatography to isolate the high-purity 3-benzylidene-2,3-dihydroquinolone product.
Commercial Advantages for Procurement and Supply Chain Teams
From a procurement and supply chain perspective, this patented methodology offers substantial strategic advantages over legacy synthesis routes. The primary benefit stems from the use of commercially available and cost-effective starting materials. N-pyridinesulfonyl-o-iodoaniline can be synthesized in high volumes from commodity chemicals like o-iodoaniline and pyridine sulfonyl chloride, ensuring a stable and resilient supply base that is not subject to the volatility of exotic reagent markets. Similarly, the allene substrates are accessible through established olefin synthesis pathways. This reliance on abundant feedstocks significantly mitigates supply chain risks and allows for long-term contracting at favorable price points, providing a competitive edge in the manufacturing of high-value pharmaceutical intermediates.
- Cost Reduction in Manufacturing: The economic viability of this process is driven by the elimination of expensive and hazardous reagents such as gaseous carbon monoxide and high-pressure reactor setups. By replacing these with a solid CO surrogate and operating at atmospheric pressure in standard glass-lined or stainless steel reactors, capital expenditure (CAPEX) and operational expenditure (OPEX) are drastically reduced. Furthermore, the high reaction yields (often >85%) minimize raw material waste and reduce the load on downstream purification units. The simplified workup procedure, which involves basic filtration and standard chromatography rather than complex distillations or crystallizations, lowers energy consumption and labor costs, contributing to a leaner and more profitable manufacturing model.
- Enhanced Supply Chain Reliability: The robustness of the reaction conditions translates directly into improved supply chain reliability. Because the process tolerates a wide range of functional groups and impurities in the starting materials better than sensitive alternative methods, the risk of batch failure due to raw material variability is minimized. The use of toluene, a ubiquitous industrial solvent, ensures that solvent supply is never a bottleneck. Additionally, the relatively short reaction time of 24 to 48 hours allows for faster turnaround times between batches, enabling manufacturers to respond more agilely to fluctuating market demands and urgent customer orders without the need for excessive inventory buffering.
- Scalability and Environmental Compliance: Scaling this process from gram to kilogram or tonne levels is straightforward due to the absence of exothermic hazards associated with gas handling and the use of common solvents. The reaction generates minimal hazardous waste, aligning with increasingly stringent environmental regulations and corporate sustainability mandates. The solid byproduct from the CO surrogate decomposition can be easily filtered off, simplifying waste treatment protocols. This environmental friendliness not only reduces disposal costs but also enhances the marketability of the final product to eco-conscious pharmaceutical clients who prioritize green chemistry metrics in their supplier selection criteria.
Frequently Asked Questions (FAQ)
The following questions address common technical inquiries regarding the implementation and optimization of this palladium-catalyzed synthesis. These answers are derived directly from the experimental data and mechanistic understanding presented in the patent documentation, providing clarity for process engineers and quality assurance teams evaluating this technology for adoption. Understanding these nuances is critical for ensuring consistent product quality and maximizing the efficiency of the manufacturing process.
Q: What is the optimal temperature and time for this carbonylation reaction?
A: According to the patented process, the reaction is optimally conducted at 90°C for a duration of 24 to 48 hours. This thermal window ensures sufficient energy for the oxidative addition and subsequent migratory insertion steps while maintaining substrate stability.
Q: Which solvents and additives are critical for high yield?
A: Toluene is the preferred organic solvent due to its ability to dissolve reactants effectively and support high conversion rates. Triethylamine serves as a crucial additive (2.0 equivalents) to facilitate the reaction progress, while TFBen acts as a safe carbon monoxide substitute.
Q: Does this method tolerate diverse functional groups on the allene substrate?
A: Yes, the methodology exhibits excellent substrate compatibility. It successfully accommodates various substituents on the aryl ring of the allene, including electron-donating groups like methyl and methoxy, as well as electron-withdrawing halogens such as fluorine, chlorine, and bromine, without significant loss in yield.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 3-Benzylidene-2,3-dihydroquinolone Supplier
At NINGBO INNO PHARMCHEM, we recognize the transformative potential of this advanced palladium-catalyzed carbonylation technology for the pharmaceutical industry. As a premier CDMO partner, we possess the technical expertise and infrastructure to translate this patented laboratory method into a robust, commercial-scale manufacturing process. Our team has extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your supply needs are met with unwavering consistency. We operate stringent purity specifications and utilize rigorous QC labs equipped with state-of-the-art analytical instrumentation to guarantee that every batch of 3-benzylidene-2,3-dihydroquinolone intermediate meets the highest global standards for potency and impurity profiles.
We invite you to collaborate with us to leverage this cutting-edge synthesis for your drug development programs. By partnering with our technical procurement team, you can access a Customized Cost-Saving Analysis tailored to your specific volume requirements and timeline. We encourage potential partners to contact us directly to request specific COA data for our catalog compounds or to discuss route feasibility assessments for novel analogs. Let us help you accelerate your pipeline with reliable, cost-effective, and high-quality chemical solutions that drive innovation in analgesic and antitumor therapeutics.