Scalable Synthesis of Novel Ursolic Acid Pyrimidine Amide Derivatives for Oncology Applications
Scalable Synthesis of Novel Ursolic Acid Pyrimidine Amide Derivatives for Oncology Applications
The pharmaceutical industry is constantly seeking novel scaffolds that can offer improved therapeutic indices for oncology treatments. Patent CN113004368B discloses a groundbreaking series of ursolic acid pyrimidine amide derivatives, representing a significant advancement in the field of medicinal chemistry and organic synthesis. These compounds are designed by strategically modifying the natural triterpenoid backbone of ursolic acid, a compound known for its wide distribution in nature and low acquisition cost. The innovation lies in the fusion of a pyrimidine heterocyclic system onto the ursolic acid skeleton, creating a hybrid molecule that exhibits potent inhibitory effects against human lung cancer (A549), breast cancer (MCF-7), cervical cancer (HeLa), and liver cancer (Hep G2) cell lines. This structural modification not only enhances biological activity but also maintains low toxicity towards normal human liver epithelial cells (Ges-1), addressing a critical safety concern in chemotherapy drug development.
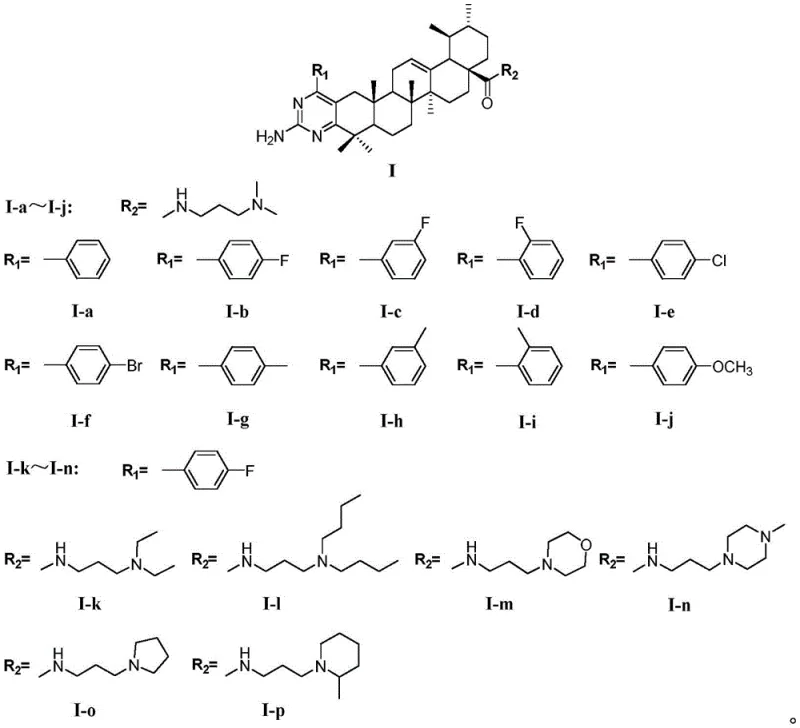
For R&D directors and procurement specialists looking for a reliable pharmaceutical intermediate supplier, this technology offers a robust pathway to high-value antitumor agents. The synthesis strategy leverages the inherent reactivity of the ursolic acid carboxyl group and the C-3 position, allowing for diverse derivatization. By introducing different substituents on the benzylidene moiety and varying the amine side chains, a comprehensive library of derivatives can be generated to optimize structure-activity relationships (SAR). This flexibility is crucial for lead optimization campaigns in early-stage drug discovery, providing a versatile platform for developing next-generation anticancer therapeutics with enhanced efficacy and safety profiles.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditional approaches to modifying ursolic acid often focus on simple esterification or etherification at the C-3 hydroxyl group or the C-28 carboxyl group. While these methods are straightforward, they frequently result in derivatives with marginal improvements in solubility or bioavailability, failing to unlock the full potential of the triterpenoid scaffold. Many conventional derivatives suffer from poor water solubility and limited cellular uptake, which restricts their clinical utility. Furthermore, simple modifications often lack the specific molecular interactions required to effectively inhibit tumor cell proliferation, leading to compounds with weak cytotoxicity that cannot compete with established chemotherapeutic agents. The lack of nitrogen-containing heterocycles in these traditional structures limits their ability to form hydrogen bonds with biological targets, a key factor in achieving high binding affinity and selectivity.
The Novel Approach
The novel approach detailed in the patent overcomes these limitations by constructing a complex pyrimidine-fused system directly onto the ursolic acid core. This method involves a multi-step sequence that transforms the A-ring and the side chain simultaneously, creating a rigidified structure that mimics known pharmacophores found in successful anticancer drugs. By utilizing a Claisen-Schmidt condensation followed by amidation and cyclization, the process introduces a planar heterocyclic ring that can intercalate with DNA or interact with enzyme active sites more effectively than the flexible aliphatic chains of conventional derivatives. This strategic design results in compounds like I-b and I-h, which demonstrate superior IC50 values compared to the positive control etoposide in certain cell lines. The approach effectively merges the anti-inflammatory and antioxidant properties of ursolic acid with the antiproliferative capabilities of pyrimidine derivatives, yielding a synergistic effect that is highly desirable in modern oncology drug design.
Mechanistic Insights into the Multi-Step Synthetic Route
The synthesis of these high-purity pharmaceutical intermediates relies on a precise four-step cascade that ensures high regioselectivity and yield. The process begins with the oxidation of the C-3 hydroxyl group on the A-ring of ursolic acid. Using Jones reagent (chromic acid in sulfuric acid), the secondary alcohol is converted into a ketone, yielding 3-oxoursolic acid. This oxidation is critical as it activates the alpha-position for subsequent condensation reactions. The reaction conditions must be carefully controlled, typically involving an ice-water bath initially to manage the exotherm, followed by stirring at room temperature to ensure complete conversion. The resulting 3-oxo intermediate serves as the electrophilic partner for the next stage.
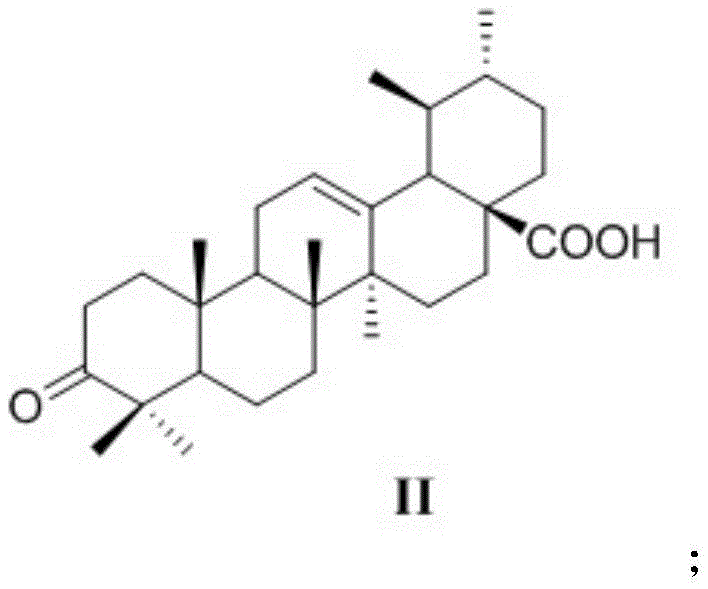
Following oxidation, the 3-oxoursolic acid undergoes a Claisen-Schmidt condensation with various substituted benzaldehydes. Under alkaline conditions provided by potassium hydroxide in ethanol, the enolate formed at the C-2 position attacks the carbonyl carbon of the benzaldehyde. This step introduces the aromatic diversity essential for SAR studies, allowing for the incorporation of electron-withdrawing groups like fluorine or chlorine, as well as electron-donating groups like methyl or methoxy. The reaction proceeds to form a benzylidene double bond conjugated with the ketone, extending the pi-system of the molecule. This conjugation is vital for the electronic properties of the final drug candidate, influencing its redox potential and interaction with biological macromolecules. The crude product is typically purified by extraction and recrystallization to remove unreacted aldehydes and polymeric byproducts.
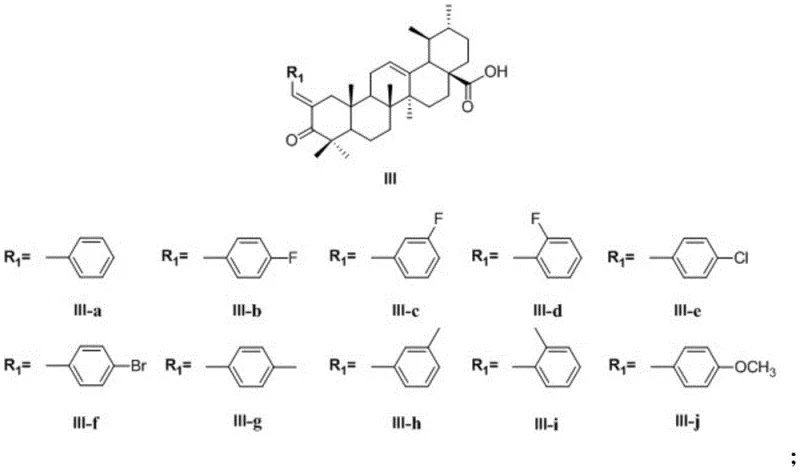
The third stage involves the activation of the C-28 carboxyl group to form an amide bond. This is achieved using standard peptide coupling reagents, specifically DCC (dicyclohexylcarbodiimide) and HOBt (1-hydroxybenzotriazole), in dichloromethane. The use of HOBt suppresses racemization and improves coupling efficiency, ensuring that the chiral integrity of the ursolic acid backbone is preserved. Various amines, such as 3-dimethylamino-1-propylamine or cyclic amines like morpholine and piperazine derivatives, are introduced at this stage. This step installs the basic side chain that improves the water solubility of the final compound and provides a handle for salt formation, which is often necessary for formulation development. The reaction mixture is stirred overnight at room temperature to ensure complete consumption of the expensive triterpenoid intermediate.
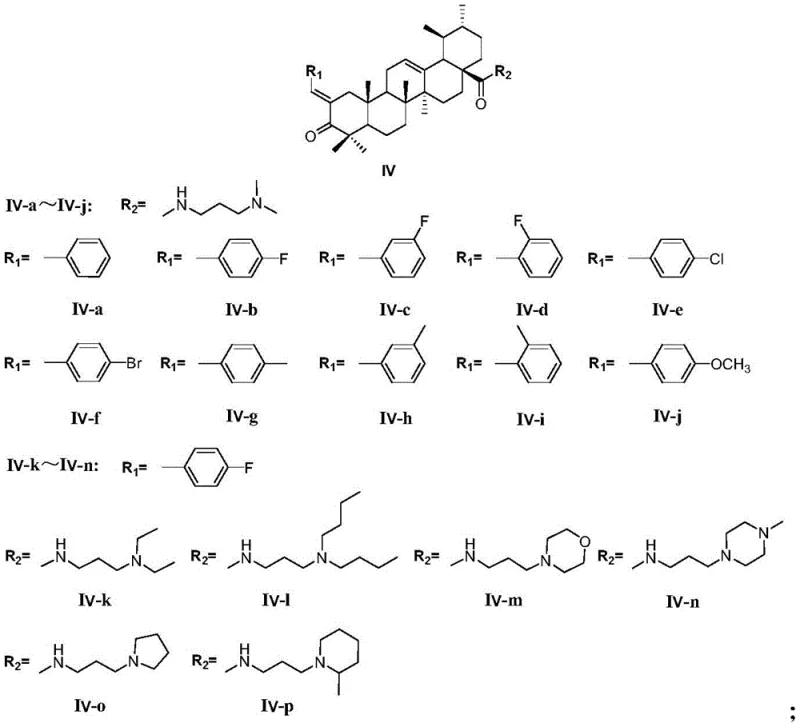
The final transformation is the construction of the pyrimidine ring. The benzylidene ursolic acid amide intermediate reacts with guanidine hydrochloride in the presence of a strong base, potassium tert-butoxide, in tert-butyl alcohol. Heating the mixture to reflux facilitates the cyclocondensation reaction, where the guanidine nitrogen attacks the electrophilic centers of the enone system, closing the ring to form the 2-aminopyrimidine moiety. This step requires rigorous temperature control and extended reaction times, typically around 24 hours, to drive the equilibrium towards the product. The final purification involves aluminum oxide column chromatography, which effectively separates the target pyrimidine derivative from urea byproducts and unreacted starting materials, delivering the high-purity API intermediate required for preclinical testing.
How to Synthesize Ursolic Acid Pyrimidine Amide Efficiently
Executing this synthesis on a commercial scale requires strict adherence to the optimized protocols described in the patent to maximize yield and minimize impurities. The process is designed to be modular, allowing manufacturers to swap out benzaldehyde and amine components without altering the core reaction conditions significantly. Detailed standardized operating procedures for each step, including precise stoichiometry, temperature ramps, and workup sequences, are essential for reproducibility. For a comprehensive guide on the specific reaction parameters and purification techniques, please refer to the technical documentation below.
- Oxidize ursolic acid at the C-3 position using Jones reagent to form 3-oxoursolic acid.
- Perform Claisen-Schmidt condensation with substituted benzaldehydes under alkaline conditions to generate benzylidene intermediates.
- Activate the carboxyl group using DCC/HOBt and react with various amines to form amide bonds.
- Cyclize the amide intermediate with guanidine hydrochloride under basic conditions to form the final pyrimidine ring.
Commercial Advantages for Procurement and Supply Chain Teams
From a supply chain perspective, the production of ursolic acid pyrimidine amide derivatives offers distinct logistical and economic benefits. The primary starting material, ursolic acid, is a natural product extracted from abundant plant sources such as apple peels and basil, ensuring a stable and sustainable supply chain that is not subject to the volatility of petrochemical feedstocks. This natural origin aligns with the growing industry demand for green and sustainable chemistry practices. Furthermore, the synthetic route utilizes commodity chemicals for the derivatization steps, such as substituted benzaldehydes and common amines, which are readily available from multiple global suppliers, reducing the risk of single-source dependency and enhancing supply security for large-scale manufacturing campaigns.
- Cost Reduction in Manufacturing: The synthetic pathway avoids the use of precious metal catalysts like palladium or platinum, which are often cost-prohibitive and require complex removal steps to meet regulatory limits for residual metals in pharmaceuticals. Instead, the process relies on inexpensive reagents like Jones reagent and potassium hydroxide. Although chromium waste management is necessary, the overall reagent cost is significantly lower compared to transition-metal catalyzed cross-coupling reactions. Additionally, the high yields reported in the examples, particularly for the final cyclization step, contribute to a favorable cost of goods sold (COGS), making these derivatives economically viable candidates for further development.
- Enhanced Supply Chain Reliability: The robustness of the four-step synthesis allows for flexible production scheduling. Since the intermediates (3-oxoursolic acid and benzylidene derivatives) are stable solids, they can be stockpiled as strategic reserves, decoupling the production of the final API intermediate from the immediate availability of all raw materials. This inventory buffering capability is crucial for mitigating supply disruptions. Moreover, the purification methods described, such as recrystallization and standard column chromatography, are well-established unit operations in the fine chemical industry, meaning that existing manufacturing infrastructure can be utilized without the need for specialized equipment investments.
- Scalability and Environmental Compliance: While the use of Jones reagent generates chromium waste, the process is amenable to waste stream optimization and recycling protocols standard in modern chemical plants. The solvent systems employed, primarily ethanol, dichloromethane, and tert-butyl alcohol, are common industrial solvents with established recovery and distillation protocols. The ability to scale this synthesis from gram quantities in the lab to kilogram or ton scales is supported by the use of homogeneous reaction conditions that do not suffer from mass transfer limitations often seen in heterogeneous catalysis. This scalability ensures that the supply can grow in tandem with clinical demand, supporting the transition from preclinical research to commercial production without process redesign.
Frequently Asked Questions (FAQ)
Understanding the technical nuances of this synthesis is vital for partners considering collaboration. The following questions address common inquiries regarding the stability, regulatory status, and customization potential of these derivatives. Our technical team has compiled these answers based on the specific data provided in the patent literature to ensure transparency and accuracy for our clients.
Q: What is the primary advantage of introducing a pyrimidine ring to ursolic acid?
A: Introducing a pyrimidine ring significantly enhances the biological activity, specifically improving antitumor potency against cell lines like HeLa and MCF-7 compared to the parent ursolic acid structure.
Q: Are the starting materials for this synthesis commercially available?
A: Yes, ursolic acid is a widely distributed natural product that is low-cost and easy to acquire, while the substituted benzaldehydes and amines are standard commodity chemicals.
Q: How is purity controlled during the multi-step synthesis?
A: Purity is maintained through rigorous workup procedures including recrystallization, solvent extraction, and final purification using aluminum oxide column chromatography.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Ursolic Acid Pyrimidine Amide Supplier
As a leader in the fine chemical sector, NINGBO INNO PHARMCHEM is uniquely positioned to support the development of these promising antitumor agents. We possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project can seamlessly transition from benchtop discovery to market supply. Our state-of-the-art facilities are equipped with rigorous QC labs capable of meeting stringent purity specifications required for pharmaceutical intermediates. We understand the critical nature of oncology drug development and are committed to providing consistent quality and reliable delivery schedules to keep your pipeline moving forward.
We invite you to engage with our technical procurement team to discuss how we can tailor our manufacturing capabilities to your specific needs. Whether you require a Customized Cost-Saving Analysis for a specific analog or need to obtain specific COA data and route feasibility assessments for a novel derivative, we are ready to assist. Contact us today to explore how our expertise in heterocyclic synthesis and natural product modification can accelerate your drug discovery programs and reduce your time to market.