Advanced Synthesis of Soluble Pyrrolopyrimidine Derivatives for Next-Generation JAK Inhibitors
Advanced Synthesis of Soluble Pyrrolopyrimidine Derivatives for Next-Generation JAK Inhibitors
The pharmaceutical industry is constantly seeking to optimize the physicochemical properties of kinase inhibitors to enhance their clinical efficacy and patient compliance. A significant breakthrough in this domain is documented in patent CN111087412B, which discloses a novel class of pyrrolopyrimidine derivatives designed to overcome the solubility limitations of earlier generation JAK inhibitors. Specifically, this technology addresses the poor aqueous solubility of the reference compound WXFL10203614, a known JAK inhibitor used for treating autoimmune diseases like rheumatoid arthritis. By introducing specific structural modifications, particularly at the nitrogen position of the pyrrole ring and utilizing a fused piperidine-triazole side chain, the inventors have achieved a dramatic improvement in water solubility without compromising the core pharmacophore required for kinase inhibition. This development represents a critical advancement for reliable pyrrolopyrimidine derivative suppliers aiming to support the next wave of immunomodulatory therapies.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Prior art methods for synthesizing JAK inhibitors often resulted in compounds with suboptimal biopharmaceutical profiles. The reference compound WXFL10203614, while potent, suffers from extremely low water solubility (0.12 mg/L), which severely limits its oral bioavailability and necessitates complex formulation strategies or high dosing regimens. From a synthetic chemistry perspective, traditional routes to similar heterocyclic scaffolds frequently involve harsh reaction conditions, expensive transition metal catalysts, or multi-step sequences with poor atom economy. These factors contribute to high manufacturing costs and generate significant chemical waste, posing challenges for cost reduction in pharmaceutical intermediate manufacturing. Furthermore, the purification of such complex molecules often requires extensive chromatography, which is difficult to translate from laboratory bench scale to industrial production without incurring substantial yield losses.
The Novel Approach
The methodology outlined in CN111087412B offers a streamlined, convergent synthetic strategy that directly addresses these inefficiencies. The core innovation lies in the modular assembly of the molecule, separating the synthesis of the complex amine side chain (Intermediate 6) from the functionalization of the pyrrolopyrimidine core. This approach allows for the independent optimization of each fragment before the final coupling step. The process utilizes readily available starting materials and avoids the use of precious metal catalysts, relying instead on classical organic transformations such as nucleophilic substitution, amidation, and hydrazine-mediated cyclization. By employing mild reaction conditions, such as room temperature sulfonylation and aqueous ethanol reflux for the final ring closure, the process ensures high purity and minimizes the formation of difficult-to-remove impurities. This robust synthetic design facilitates the commercial scale-up of complex heterocyclic compounds while maintaining strict control over the impurity profile.
Mechanistic Insights into Convergent Heterocyclic Assembly
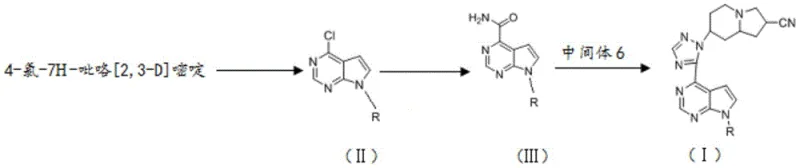
The synthesis of the key building block, Intermediate 6, involves a fascinating cascade of transformations starting from 7-amino-5,6,7,8-tetrahydroimidazo[1,2-a]pyridine-2-carboxylic acid ethyl ester. The sequence begins with the reduction of the ester to an alcohol using lithium aluminum hydride, followed by oxidation to an aldehyde using manganese dioxide. Subsequent reaction with hydroxylamine yields an oxime, which undergoes dehydration and cyclization in acetic anhydride to form the nitrile-containing fused ring system. The critical step involves the reaction with hydrazine hydrate to generate the hydrazine intermediate, which finally cyclizes with formamide to produce the triazole-fused piperidine structure of Intermediate 6. This multi-step sequence is highly efficient, converting a simple bicyclic precursor into a complex tricyclic amine with high stereochemical integrity.
The final coupling reaction between Intermediate 6 and the pyrrolopyrimidine intermediates (such as Intermediate 7, 9, 11, or 13) proceeds via a nucleophilic attack followed by cyclization. In the case of the unsubstituted core (Intermediate 7), the primary amine of the hydrazine moiety attacks the nitrile or amidine carbon, facilitated by the thermal energy of refluxing ethanol and water. For the sulfonyl-substituted variants (Compounds 2, 3, and 4), the electron-withdrawing nature of the sulfonyl group at the N-7 position of the pyrrole ring modulates the electronic density of the heterocycle, potentially enhancing the reactivity towards nucleophilic attack or stabilizing the transition state. The use of an ethanol/water mixture as the solvent system is particularly advantageous; it provides sufficient solubility for the polar intermediates while allowing for the easy removal of solvent post-reaction. This mechanistic understanding is vital for reducing lead time for high-purity kinase inhibitors by enabling precise control over reaction endpoints and crystallization processes.
How to Synthesize Pyrrolopyrimidine Derivatives Efficiently
The practical execution of this synthesis requires careful attention to stoichiometry and purification protocols to ensure the high quality demanded by pharmaceutical applications. The process is divided into two main stages: the preparation of the fused piperidine-triazole amine (Intermediate 6) and the subsequent coupling with the functionalized pyrrolopyrimidine core. Each step has been optimized in the patent examples to maximize yield and minimize byproduct formation, utilizing standard workup procedures like extraction, washing, and silica gel chromatography. For industrial implementation, the chromatography steps can often be replaced by crystallization or trituration to improve throughput. The detailed standardized synthetic steps for producing these high-value intermediates are outlined below.
- Synthesize Intermediate 6 starting from 7-amino-5,6,7,8-tetrahydroimidazo[1,2-a]pyridine-2-carboxylic acid ethyl ester through reduction, oxidation, oxime formation, cyclization, and hydrazinolysis.
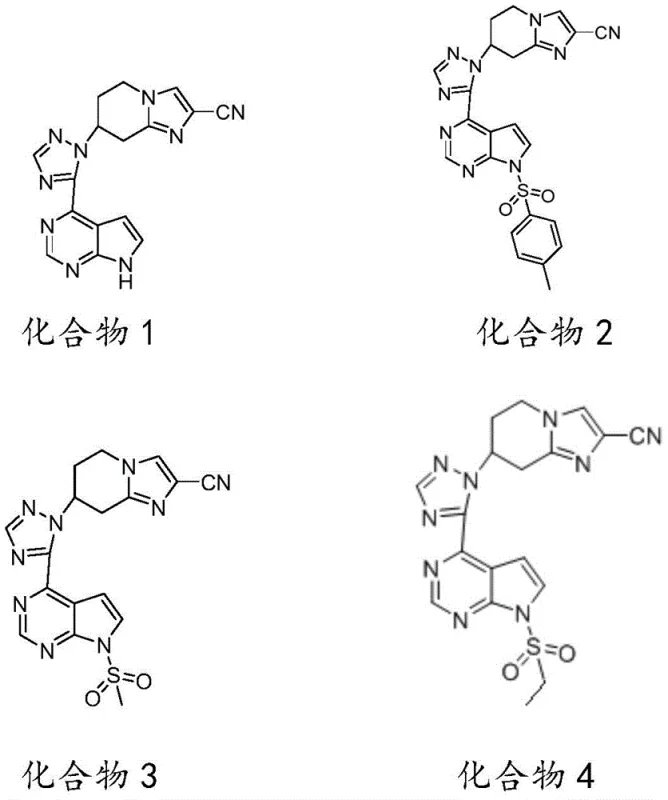
- Prepare the pyrrolopyrimidine core (Intermediate 7, 9, 11, or 13) by reacting 4-chloro-7H-pyrrolo[2,3-d]pyrimidine with formamide or sulfonyl chlorides followed by amidation.
- Perform the final ring-closure condensation by refluxing Intermediate 6 with the pyrrolopyrimidine intermediate in an ethanol/water mixture to yield the target compound.
Commercial Advantages for Procurement and Supply Chain Teams
From a procurement and supply chain perspective, the synthetic route described in CN111087412B offers compelling advantages over alternative pathways for generating JAK inhibitor scaffolds. The reliance on commodity chemicals and the avoidance of specialized reagents significantly de-risk the supply chain. The ability to produce compounds with vastly superior solubility profiles also translates to downstream benefits for drug product manufacturers, potentially reducing the need for expensive solubilizing excipients or complex particle engineering. This holistic view of value creation extends beyond mere synthesis cost to encompass the total cost of goods for the final drug product.
- Cost Reduction in Manufacturing: The synthetic route eliminates the need for expensive transition metal catalysts (such as palladium or rhodium) which are often required for cross-coupling reactions in similar heterocyclic syntheses. By utilizing base-mediated substitutions and thermal cyclizations, the process drastically reduces raw material costs and removes the necessity for costly heavy metal scavenging steps to meet regulatory limits. Furthermore, the use of common solvents like ethanol, water, and dichloromethane allows for efficient solvent recovery and recycling, contributing to substantial operational expenditure savings.
- Enhanced Supply Chain Reliability: The starting material, 4-chloro-7H-pyrrolo[2,3-d]pyrimidine, is a commercially available bulk chemical (CAS 3680-69-1), ensuring a stable and continuous supply without reliance on custom synthesis for the core scaffold. The modular nature of the synthesis means that different analogs (Compounds 1-4) can be produced from a common pool of Intermediate 6, allowing manufacturers to respond flexibly to changing demand for specific derivatives without retooling the entire production line. This flexibility is crucial for maintaining continuity of supply in the fast-paced pharmaceutical market.
- Scalability and Environmental Compliance: The reaction conditions are inherently scalable, operating at atmospheric pressure and moderate temperatures (room temperature to reflux). The final purification steps described involve silica gel chromatography, which in a commercial setting can be adapted to large-scale column chromatography or, preferably, optimized for crystallization to reduce solvent waste. The aqueous workups and the use of less hazardous reagents align well with green chemistry principles, simplifying waste treatment and ensuring compliance with increasingly stringent environmental regulations regarding solvent discharge and heavy metal content.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of these pyrrolopyrimidine derivatives. The answers are derived directly from the experimental data and technical specifications provided in the patent literature, offering a transparent view of the technology's capabilities and limitations for potential partners and licensees.
Q: How does the water solubility of these new derivatives compare to the prior art compound WXFL10203614?
A: According to patent CN111087412B, the new derivatives exhibit significantly improved water solubility. While WXFL10203614 has a solubility of only 0.12 mg/L, Compound 2 reaches 2.87 mg/L, representing a massive improvement for bioavailability and formulation.
Q: What are the key raw materials required for this synthesis?
A: The synthesis relies on commercially available starting materials such as 4-chloro-7H-pyrrolo[2,3-d]pyrimidine (CAS 3680-69-1) and 7-amino-5,6,7,8-tetrahydroimidazo[1,2-a]pyridine-2-carboxylic acid ethyl ester, ensuring a stable supply chain.
Q: Is this process suitable for large-scale commercial production?
A: Yes, the process utilizes standard solvents like ethanol, water, THF, and DCM, and avoids exotic catalysts. The reactions operate at manageable temperatures (room temperature to reflux), making it highly amenable to kilogram-to-ton scale-up.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Pyrrolopyrimidine Derivative Supplier
The technological potential of the synthesis route disclosed in CN111087412B is immense, offering a clear path to high-solubility JAK inhibitors that could redefine treatment standards for autoimmune disorders. At NINGBO INNO PHARMCHEM, we possess the technical expertise and infrastructure to translate this patented chemistry from the laboratory bench to commercial reality. Our team has extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project moves seamlessly from clinical trials to market launch. We maintain stringent purity specifications and operate rigorous QC labs equipped with state-of-the-art analytical instrumentation to guarantee that every batch of intermediate meets the exacting standards required for API synthesis.
We invite you to collaborate with us to optimize this supply chain and unlock the full value of this innovative chemistry. Contact our technical procurement team today to request a Customized Cost-Saving Analysis tailored to your specific volume requirements. We are ready to provide specific COA data and comprehensive route feasibility assessments to demonstrate how our manufacturing capabilities can accelerate your development timeline and reduce your overall cost of goods.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →