Scalable Synthesis of D-2-Aminooxy-3-Methylbutyric Acid for Advanced Pharmaceutical Intermediates
The pharmaceutical and fine chemical industries are constantly seeking robust pathways to access non-natural amino acid derivatives that serve as critical building blocks for novel therapeutic agents. Patent CN102911085A introduces a sophisticated and highly selective synthesis process for D-2-aminooxy-3-methylbutyric acid, a compound of significant interest for the design of chiral shift reagents and pseudo-peptide structures. This specific molecule replaces a carbon atom in the natural amino acid skeleton with an oxygen atom, creating a unique right-hand α N-O corner that exhibits distinct physiological activity and resistance to enzymatic degradation within the body. The technical breakthrough lies in the strategic utilization of natural L-valine as the chiral pool starting material, which inherently reduces the complexity of stereochemical control compared to racemic synthesis. By leveraging a sequence of diazotization hydrolysis, acetyl protection, and a pivotal Mitsunobu reaction, the process achieves high reaction selectivity and yield under mild conditions. This approach not only addresses the scarcity of optically active raw materials often cited in prior art but also establishes a reliable foundation for the industrial manufacturing of high-purity pharmaceutical intermediates. 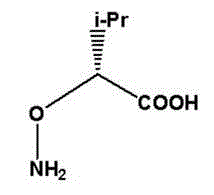
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of D-aminooxy acid compounds has been plagued by significant technical hurdles that hinder efficient commercial production. Traditional methods often rely on the substitution reaction of 2-halogenated acids with AcNHOK or the reaction of 2-diazotic acids with acyl hydroxylamine compounds, as documented in earlier chemical literature. These conventional pathways frequently suffer from poor control over the chirality of the final product, leading to racemic mixtures that require expensive and yield-reducing resolution steps. Furthermore, many existing methods depend on optically active raw materials that are not easily obtainable on a large scale, creating supply chain bottlenecks and driving up costs for downstream manufacturers. The lack of stereoselectivity in these older reactions means that significant resources are wasted on separating enantiomers, and the harsh reaction conditions often associated with halogenated intermediates pose safety and environmental challenges. Consequently, the industry has long needed a method that guarantees high optical purity without relying on scarce or prohibitively expensive chiral precursors.
The Novel Approach
The methodology outlined in CN102911085A represents a paradigm shift by utilizing abundant natural L-valine to access the non-natural D-configuration through a controlled inversion strategy. This novel approach bypasses the need for difficult-to-source chiral reagents by starting with a cheap, renewable amino acid and chemically transforming it through a well-defined seven-step sequence. The process is characterized by mild reaction conditions that are easy to control, such as maintaining temperatures between 0°C and 65°C, which significantly reduces energy consumption and operational risks in a plant setting. By employing a Mitsunobu reaction for the critical stereochemical inversion, the synthesis ensures high selectivity, effectively minimizing the formation of unwanted by-products and simplifying the purification workflow. The use of common organic solvents like ethyl acetate, methanol, and dichloromethane further enhances the feasibility of this route for large-scale operations, as these materials are readily available and easy to recover. This combination of accessible raw materials, high selectivity, and operational simplicity makes the process exceptionally suitable for industrial production, offering a clear advantage over legacy methods.
Mechanistic Insights into Mitsunobu Inversion and Diazotization
The core of this synthesis strategy relies on two critical chemical transformations that dictate the overall efficiency and stereochemical outcome of the process. The initial step involves the diazotization hydrolysis of L-valine using sodium nitrite and sulfuric acid in an aqueous medium, which converts the amino group into a hydroxyl group while retaining the chiral center to form (S)-2-hydroxy-3-methylbutyric acid. This step is crucial as it sets the stage for the subsequent inversion, requiring precise temperature control between 0°C and 5°C to prevent racemization and ensure high conversion rates. Following protection of the hydroxyl group via acetylation and esterification to form the tert-butyl ester, the synthesis reaches its pivotal moment with the Mitsunobu reaction. In this step, the (S)-hydroxy intermediate reacts with N-hydroxyphthalimide in the presence of triphenylphosphine and diethylazodicarboxylate (DEAD) in tetrahydrofuran. This reaction mechanism facilitates a nucleophilic substitution with inversion of configuration, effectively converting the (S)-alcohol into the (R)-aminooxy derivative with high fidelity. The use of the phthalimide group serves as a robust protecting group for the aminooxy functionality, allowing for subsequent manipulation without compromising the newly established stereocenter.
Impurity control is meticulously managed throughout the synthesis through a combination of selective reactivity and strategic protection group chemistry. The acetyl protection in the early stages prevents unwanted side reactions at the hydroxyl group during esterification, while the tert-butyl ester protects the carboxylic acid from participating in the Mitsunobu coupling. The deprotection steps, utilizing potassium carbonate in a methanol-water mixture for the phthalimide group and trifluoroacetic acid for the tert-butyl ester, are designed to be highly specific, minimizing the risk of hydrolyzing other sensitive bonds within the molecule. The final purification via silica gel column chromatography and ethanol recrystallization ensures that the target D-2-aminooxy-3-methylbutyric acid meets stringent purity specifications required for pharmaceutical applications. By avoiding heavy metal catalysts and utilizing organic reagents that can be effectively removed during workup, the process inherently limits the introduction of toxic impurities. This attention to mechanistic detail ensures that the final product possesses the high optical purity necessary for its intended use in chiral shift reagents and bioactive peptide design.
How to Synthesize D-2-Aminooxy-3-Methylbutyric Acid Efficiently
Executing this synthesis requires a disciplined approach to reaction conditions and stoichiometry to maximize the overall yield, which the patent reports at approximately 8.4% over seven steps. The process begins with the careful addition of sodium nitrite to an acidic solution of valine, followed by a controlled warm-up to room temperature to complete the diazotization. Subsequent steps involve standard organic transformations such as acylation with acetyl chloride and esterification using DCC and DMAP, which are well-understood in process chemistry. The critical Mitsunobu step demands anhydrous conditions and precise molar ratios of DEAD and triphenylphosphine to ensure complete inversion without excessive reagent waste. The final deprotection and hydrolysis steps utilize mild basic and acidic conditions respectively, allowing for the gentle release of the free acid without degrading the sensitive aminooxy linkage. 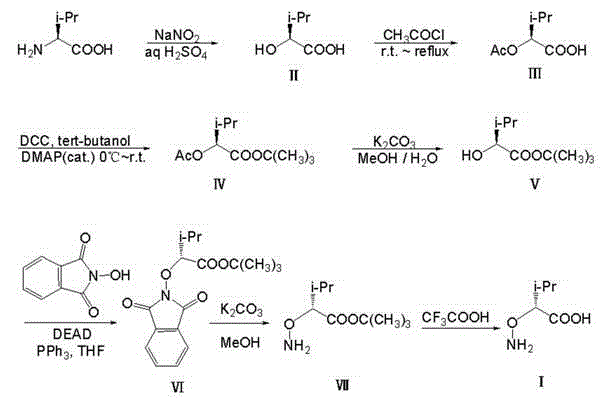
- Convert L-Valine to (S)-2-hydroxy-3-methylbutyric acid via diazotization hydrolysis using sodium nitrite and sulfuric acid at low temperatures.
- Protect the hydroxyl group via acetylation and form the tert-butyl ester using DCC and DMAP catalysis.
- Perform Mitsunobu reaction with N-hydroxyphthalimide to invert stereochemistry, followed by deprotection and hydrolysis to yield the target acid.
Commercial Advantages for Procurement and Supply Chain Teams
From a procurement and supply chain perspective, this synthesis route offers substantial strategic benefits by leveraging widely available commodity chemicals as starting materials. The reliance on L-valine, a natural amino acid produced in massive quantities globally, insulates the supply chain from the volatility often associated with specialized chiral reagents. This accessibility translates directly into enhanced supply chain reliability, as manufacturers are not dependent on single-source suppliers for exotic starting materials that could lead to production delays. Furthermore, the mild reaction conditions and the use of standard solvents reduce the need for specialized high-pressure or high-temperature equipment, lowering the barrier to entry for contract manufacturing organizations. The elimination of complex resolution steps typically required for racemic syntheses significantly streamlines the production timeline, allowing for faster turnaround times on custom orders. By simplifying the purification process through high-selectivity reactions, the overall consumption of chromatography media and solvents is reduced, contributing to a more sustainable and cost-efficient manufacturing footprint.
- Cost Reduction in Manufacturing: The economic viability of this process is driven by the use of low-cost, natural L-valine as the primary feedstock, which eliminates the premium pricing associated with synthetic chiral building blocks. By achieving high stereoselectivity through the Mitsunobu inversion, the process avoids the significant material losses inherent in chiral resolution techniques, thereby maximizing the yield of the desired enantiomer per batch. The avoidance of expensive transition metal catalysts further reduces raw material costs and simplifies the waste treatment process, as there is no need for costly heavy metal scavenging steps. Additionally, the ability to perform multiple steps without intermediate purification, as suggested by the direct use of crude products in subsequent reactions, significantly reduces labor and solvent consumption. These factors combine to create a manufacturing profile that supports substantial cost savings without compromising on the quality of the final intermediate.
- Enhanced Supply Chain Reliability: The robustness of this synthesis route ensures consistent production output, which is critical for maintaining uninterrupted supply to downstream pharmaceutical clients. Since the reagents involved, such as sodium nitrite, acetyl chloride, and common organic solvents, are standard industrial chemicals, the risk of supply disruption is minimized compared to routes relying on bespoke reagents. The mild operating conditions also reduce the likelihood of equipment failure or safety incidents that could halt production, ensuring a steady flow of material. This reliability allows procurement managers to plan inventory levels with greater confidence, reducing the need for excessive safety stock and freeing up working capital. The scalability of the process means that supply can be rapidly ramped up to meet surges in demand, providing a flexible partner for growing drug development programs.
- Scalability and Environmental Compliance: The process is inherently designed for scale-up, utilizing reaction conditions that are easily transferable from laboratory to pilot and commercial plant scales. The use of aqueous workups and common organic solvents facilitates efficient solvent recovery and recycling, aligning with modern environmental compliance standards and reducing the overall environmental footprint. The absence of toxic heavy metals simplifies waste disposal and reduces the regulatory burden associated with effluent treatment. The high selectivity of the reactions minimizes the generation of complex by-product mixtures, making waste streams easier to treat and manage. This environmental efficiency not only supports corporate sustainability goals but also reduces the operational costs associated with waste management and regulatory compliance, making the process attractive for long-term commercial production.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of D-2-aminooxy-3-methylbutyric acid. These answers are derived directly from the technical specifications and advantages detailed in the patent literature, providing clarity on the feasibility and benefits of this synthesis route. Understanding these details is essential for R&D and procurement teams evaluating this intermediate for their specific supply chain needs. The information below highlights the balance between technical precision and commercial practicality that defines this manufacturing process.
Q: What is the primary advantage of using L-Valine as the starting material?
A: Using natural L-Valine ensures high optical purity at the start of the synthesis and leverages a widely available, cost-effective renewable feedstock compared to synthetic racemic mixtures.
Q: How is stereochemical inversion achieved in this process?
A: The process utilizes a Mitsunobu reaction with N-hydroxyphthalimide, which effectively inverts the configuration from the (S)-hydroxy intermediate to the (R)-aminooxy product with high fidelity.
Q: Is this synthesis route suitable for large-scale industrial production?
A: Yes, the patent highlights mild reaction conditions, easy-to-control temperatures, and the use of common solvents like ethyl acetate and methanol, making it highly adaptable for commercial scale-up.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable D-2-Aminooxy-3-Methylbutyric Acid Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that high-quality chiral intermediates play in the development of next-generation therapeutics and diagnostic reagents. Our technical team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the sophisticated synthesis route described in CN102911085A can be effectively translated into a robust industrial process. We are committed to delivering products that meet stringent purity specifications, supported by our rigorous QC labs that employ advanced analytical techniques to verify identity and optical purity. Our infrastructure is designed to handle complex multi-step syntheses involving sensitive reagents like those used in Mitsunobu reactions, guaranteeing consistency and reliability for our global partners. By choosing us as your manufacturing partner, you gain access to a team that understands both the chemical nuances and the commercial imperatives of the fine chemical industry.
We invite you to collaborate with us to optimize your supply chain for this valuable intermediate. Our experts are ready to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements and quality standards. We encourage potential partners to contact our technical procurement team to request specific COA data and route feasibility assessments that demonstrate our capability to meet your project timelines. Whether you are in the early stages of drug discovery or preparing for commercial launch, our flexible manufacturing solutions are designed to support your growth and ensure a secure supply of critical materials.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →