Advanced Synthesis of Varenicline Tartrate Intermediate Impurity for Global QAQC Standards
Advanced Synthesis of Varenicline Tartrate Intermediate Impurity for Global QAQC Standards
The pharmaceutical industry continuously demands higher purity standards for Active Pharmaceutical Ingredients (APIs), particularly for smoking cessation aids like Varenicline Tartrate. Patent CN114685497A, published in July 2022, introduces a groundbreaking methodology for the preparation of a specific intermediate impurity, designated as Impurity I. This technical advancement addresses a critical gap in quality control by providing a reliable synthesis route for a compound that typically arises as an unwanted byproduct during the main drug manufacturing process. By establishing a dedicated pathway to generate this impurity, manufacturers can now produce authentic reference standards essential for rigorous HPLC analysis and regulatory compliance. This innovation not only enhances the ability to detect trace contaminants but also ensures that the final therapeutic product maintains the highest safety profiles for patients worldwide.
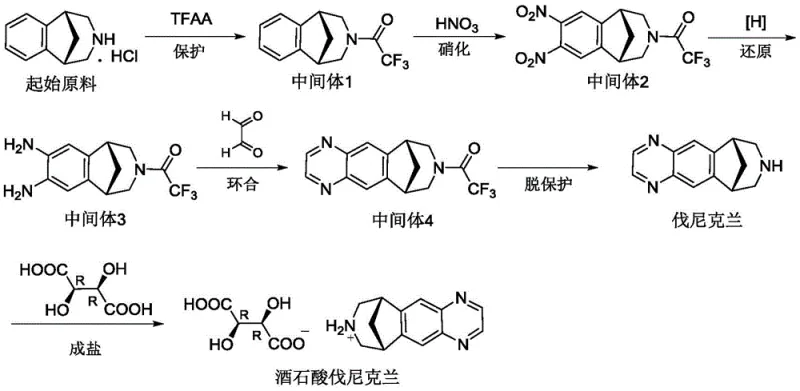
In the context of global supply chains, having access to well-characterized impurities is paramount for a reliable pharmaceutical intermediate supplier. The ability to synthesize these complex heterocyclic structures on demand allows quality assurance teams to validate their analytical methods with precision. This patent outlines a robust four-step sequence that transforms a readily available starting material into the target impurity through protection, nitration, reduction, and cyclization. The strategic value of this technology lies in its reproducibility and the high purity levels achieved, with the final product demonstrating an HPLC purity of approximately 97.36%. Such technical capabilities are indispensable for companies aiming to minimize regulatory risks and accelerate the time-to-market for generic versions of blockbuster drugs.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditionally, the management of impurities in Varenicline Tartrate synthesis has been reactive rather than proactive. In standard manufacturing routes, Impurity I is generated inadvertently during the cyclization of diamino intermediates with glyoxal or formaldehyde-containing reagents. Because this impurity forms alongside the desired product in complex mixtures, isolating it in sufficient quantity and purity for use as a reference standard is notoriously difficult. Conventional separation techniques often suffer from low recovery rates and co-elution problems, making it challenging to establish accurate limit tests. Furthermore, without a dedicated synthesis method, batch-to-batch variability in impurity profiles can lead to inconsistent quality data, complicating the validation process for regulatory submissions. This lack of control poses significant risks for cost reduction in API manufacturing, as failed batches due to unidentified peaks can result in substantial financial losses.
The Novel Approach
The methodology disclosed in patent CN114685497A represents a paradigm shift by treating the impurity as a target molecule rather than a waste product. The novel approach utilizes a linear synthetic strategy that deliberately constructs the pyrazine ring system characteristic of Impurity I. Instead of relying on the stochastic formation during the main API synthesis, this route employs triethyl orthoformate as a specific cyclizing agent under mild acidic catalysis. This controlled environment ensures that the reaction proceeds selectively towards the impurity structure, minimizing the formation of side products. The process integrates efficient workup procedures, such as methanol pulping and isopropanol recrystallization for intermediates, which streamline the purification workflow. For procurement teams, this translates to a more predictable supply of critical reference materials, facilitating better inventory management and reducing the lead time associated with sourcing rare analytical standards.
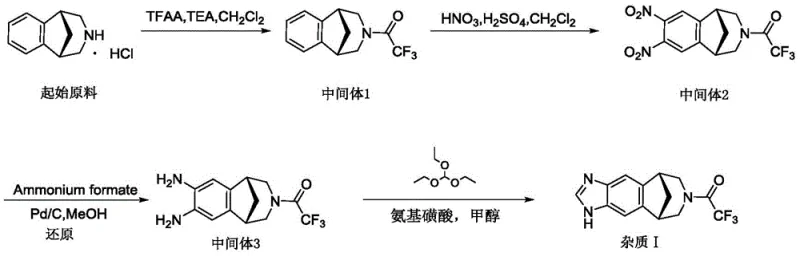
Mechanistic Insights into Sulfamic Acid-Catalyzed Cyclization
The core chemical innovation in this patent lies in the final cyclization step, where Intermediate 3 is converted into the target Impurity I. Mechanistically, this transformation involves the condensation of the ortho-diamino groups on the benzene ring with triethyl orthoformate. Sulfamic acid acts as a mild yet effective Brønsted acid catalyst, promoting the elimination of ethanol and the subsequent closure of the pyrazine ring. Unlike stronger mineral acids that might degrade the sensitive tricyclic framework or cause polymerization, sulfamic acid provides a buffered acidic environment that preserves the structural integrity of the molecule. The reaction kinetics are optimized at temperatures between 20°C and 40°C, balancing reaction rate with selectivity. This precise control over the reaction conditions is crucial for achieving the reported high purity, as it prevents the over-reaction or decomposition that often plagues heterocycle synthesis in industrial settings.
Furthermore, the preceding reduction step utilizing ammonium formate and palladium on carbon (Pd/C) offers distinct mechanistic advantages over traditional catalytic hydrogenation. This transfer hydrogenation mechanism proceeds via the decomposition of ammonium formate on the metal surface to generate active hydrogen species in situ. This avoids the mass transfer limitations associated with gaseous hydrogen diffusion into the liquid phase, leading to more uniform reduction of the dinitro intermediate. The use of methanol as a solvent further enhances the solubility of the organic intermediates while maintaining compatibility with the catalyst. From an impurity control perspective, this method minimizes the risk of over-reduction or hydrodehalogenation side reactions, ensuring that the diamino Intermediate 3 is produced with high fidelity. Such mechanistic understanding is vital for R&D directors tasked with scaling these processes from gram-scale laboratory synthesis to multi-kilogram production runs.
How to Synthesize Varenicline Impurity I Efficiently
The synthesis of this critical reference standard requires strict adherence to the reaction parameters outlined in the patent to ensure reproducibility and safety. The process begins with the protection of the amine functionality, followed by electrophilic aromatic substitution to install nitro groups, and concludes with reduction and cyclization. Each step has been optimized to maximize yield while minimizing the generation of secondary impurities. Operators must pay close attention to temperature controls, particularly during the exothermic nitration and protection steps, to prevent thermal runaways. The detailed standardized synthesis steps below provide a comprehensive guide for laboratory personnel to replicate the high-purity results described in the intellectual property documentation.
- Protect the starting amine with trifluoroacetic anhydride (TFAA) in dichloromethane using triethylamine as a base at 0-5°C to form Intermediate 1.
- Perform nitration on Intermediate 1 using fuming nitric acid and concentrated sulfuric acid at low temperature (-5°C) to introduce nitro groups, yielding Intermediate 2.
- Reduce the nitro groups of Intermediate 2 to amines using ammonium formate as a hydrogen donor and 5% Pd/C catalyst in methanol at 20-40°C to obtain Intermediate 3.
- Cyclize Intermediate 3 with triethyl orthoformate under sulfamic acid catalysis at 20-40°C, followed by pH adjustment and purification to isolate Impurity I.
Commercial Advantages for Procurement and Supply Chain Teams
Adopting the synthetic route described in this patent offers tangible benefits for the commercial viability of Varenicline Tartrate production. By internalizing the production of key impurities, pharmaceutical companies can reduce their reliance on external vendors for expensive reference standards, thereby securing their supply chain against market fluctuations. The use of commodity chemicals such as trifluoroacetic anhydride, fuming nitric acid, and ammonium formate ensures that raw material costs remain stable and predictable. Moreover, the avoidance of high-pressure hydrogenation equipment in favor of transfer hydrogenation significantly lowers the barrier to entry for manufacturing facilities that may lack specialized high-pressure reactors. This flexibility allows for a more distributed manufacturing model, enhancing overall supply chain resilience.
- Cost Reduction in Manufacturing: The implementation of transfer hydrogenation using ammonium formate eliminates the need for costly high-pressure hydrogen infrastructure and the associated safety certifications. This substitution drastically simplifies the reactor requirements, allowing the reduction step to be performed in standard glass-lined or stainless steel vessels. Additionally, the use of sulfamic acid as a catalyst is economically advantageous compared to more expensive Lewis acids or precious metal catalysts, contributing to a lower overall cost of goods sold (COGS). The streamlined workup procedures, which rely on simple extraction and crystallization rather than complex chromatographic separations for intermediates, further reduce solvent consumption and processing time.
- Enhanced Supply Chain Reliability: The starting materials and reagents specified in this route are widely available from global chemical suppliers, mitigating the risk of single-source dependency. The robustness of the synthetic steps, characterized by wide operating temperature windows and tolerance to minor variations in stoichiometry, ensures consistent output even when scaling up production volumes. This reliability is critical for maintaining continuous API manufacturing schedules and avoiding delays caused by the unavailability of specialized reagents. Furthermore, the ability to generate impurity standards in-house empowers quality control laboratories to respond rapidly to out-of-specification events without waiting for external shipments.
- Scalability and Environmental Compliance: The process design inherently supports scalability, with intermediate isolation steps like methanol pulping and isopropanol recrystallization being easily adaptable to large-scale industrial equipment. The selection of solvents such as dichloromethane and methanol allows for efficient recovery and recycling systems, aligning with modern green chemistry initiatives to reduce waste. By avoiding the use of hazardous gaseous reagents and minimizing the generation of heavy metal waste through efficient catalyst filtration, the process facilitates easier compliance with environmental regulations. This environmental stewardship not only reduces disposal costs but also enhances the corporate sustainability profile of the manufacturing entity.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the synthesis and application of Varenicline Tartrate Impurity I. These insights are derived directly from the experimental data and technical specifications provided in the patent literature. Understanding these aspects is essential for stakeholders involved in process development, quality assurance, and strategic sourcing of pharmaceutical intermediates.
Q: Why is the synthesis of Impurity I critical for Varenicline Tartrate manufacturing?
A: Impurity I is a key byproduct formed during the cyclization step of Varenicline synthesis. Having an authentic reference standard allows manufacturers to accurately quantify and control this impurity via HPLC, ensuring the final API meets stringent regulatory safety profiles.
Q: What are the advantages of using ammonium formate for reduction in this process?
A: Using ammonium formate for transfer hydrogenation eliminates the need for high-pressure hydrogen gas equipment. This significantly enhances operational safety, simplifies the reactor setup, and reduces the capital expenditure required for scaling the reduction step.
Q: How does this patent improve impurity separation compared to traditional methods?
A: The patent provides a dedicated route to synthesize Impurity I independently. This allows for better identification and separation during the main API production, preventing co-elution issues in chromatography and improving the overall purity profile of the Varenicline Tartrate.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Varenicline Impurity Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of high-purity reference standards in the development and manufacturing of complex pharmaceuticals like Varenicline Tartrate. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that we can meet the rigorous demands of global regulatory bodies. We are committed to delivering materials with stringent purity specifications, supported by our rigorous QC labs equipped with state-of-the-art analytical instrumentation. Our capability to synthesize difficult-to-access impurities allows us to support our partners in achieving flawless regulatory filings and maintaining uncompromised product quality throughout the product lifecycle.
We invite you to collaborate with us to optimize your supply chain and enhance your quality control protocols. Contact our technical procurement team today to request a Customized Cost-Saving Analysis tailored to your specific production needs. We are ready to provide specific COA data and route feasibility assessments to demonstrate how our advanced synthesis capabilities can drive efficiency and reliability in your operations. Let us be your partner in navigating the complexities of pharmaceutical intermediate manufacturing.