Advanced Synthesis of 3-Amino-Oxetane Derivatives for High-Purity Pharmaceutical Intermediates
The pharmaceutical industry continuously seeks robust and scalable pathways for constructing strained heterocyclic scaffolds, among which the 3-amino-oxetane motif has emerged as a critical pharmacophore in modern drug design. Patent CN109988126B discloses a groundbreaking methodology for the preparation of 3-amino-oxetane derivatives and their applications as key intermediates in the synthesis of bioactive molecules, including MGLUR1 receptor antagonists and non-opioid analgesics. This technology addresses the longstanding challenges associated with accessing these valuable four-membered oxygen-containing rings, offering a streamlined alternative to classical approaches. For R&D directors and procurement specialists, understanding this proprietary synthesis route is essential for securing a reliable pharmaceutical intermediate supplier capable of delivering high-purity materials with consistent quality. The disclosed method not only enhances the structural diversity available for medicinal chemistry campaigns but also provides a foundation for cost-effective manufacturing processes that align with stringent regulatory standards.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of 3-amino-oxetanes has relied on methodologies that present significant safety and economic drawbacks for large-scale operations. Traditional routes often involve the conversion of corresponding hydroxyl groups into sulfonates followed by nucleophilic displacement with sodium azide, a reagent known for its toxicity and potential explosivity, requiring specialized handling infrastructure. Another common pathway involves the formation of oximes from ketones followed by reduction, which frequently suffers from poor stereocontrol and lengthy purification sequences. Furthermore, Curtius rearrangement strategies, while effective for specific substrates, involve the generation of isocyanate intermediates that pose severe safety risks and require rigorous moisture control. These conventional approaches are characterized by long synthetic lines, expensive starting materials, and low overall atom economy, making them less attractive for the cost reduction in pharmaceutical manufacturing required by today's competitive market landscape.
The Novel Approach
In stark contrast, the methodology described in CN109988126B introduces a concise three-step sequence that circumvents the hazards of azide chemistry and the complexity of rearrangement reactions. This novel approach initiates with a direct condensation between a protected glycine ester and a ketone or aldehyde, leveraging strong bases to forge the carbon-carbon bond efficiently. The subsequent reduction and cyclization steps utilize well-established reagents that are commercially available in bulk quantities, ensuring supply chain continuity. By eliminating the need for hazardous azide intermediates and reducing the total number of isolation steps, this process significantly simplifies the operational workflow. The ability to introduce diverse substituents at the 3-position through the selection of the carbonyl partner allows for the rapid generation of analog libraries, thereby accelerating the drug discovery timeline while maintaining a favorable safety profile for production teams.
Mechanistic Insights into Base-Mediated Aldol Condensation and Cyclization
The core of this synthetic strategy lies in the initial aldol-type addition reaction, where a glycine ester derivative (Compound II) is treated with a strong non-nucleophilic base such as lithium diisopropylamide (LDA) or lithium hexamethyldisilazide (LiHMDS) at cryogenic temperatures ranging from -80°C to -5°C. This step generates a reactive enolate species which subsequently attacks the electrophilic carbonyl carbon of Compound III, forming the beta-hydroxy ester intermediate (Compound IV). The choice of base and temperature is critical for controlling the diastereoselectivity and preventing side reactions such as self-condensation of the ester. Following the formation of the carbon skeleton, the ester functionality is reduced to a primary alcohol using hydride sources like lithium aluminum hydride or sodium borohydride, yielding the 1,3-diol system (Compound V) which serves as the immediate precursor to the oxetane ring.
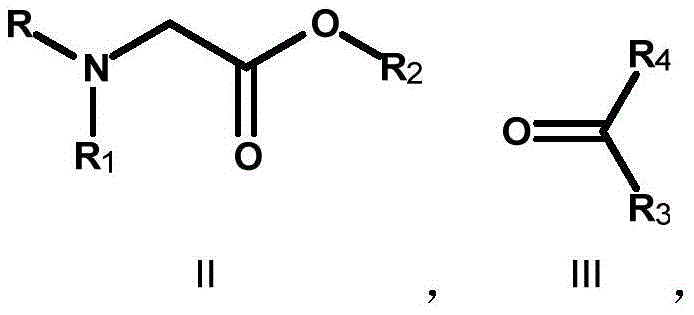
The final ring-closing step is a masterpiece of intramolecular nucleophilic substitution, where the 1,3-diol undergoes cyclization to form the strained four-membered oxetane ring (Compound VI). This transformation can be achieved through multiple mechanistic pathways depending on the specific substrate requirements. One effective method involves activation of the primary hydroxyl group with a sulfonyl chloride such as p-toluenesulfonyl chloride (TsCl) or methanesulfonyl chloride (MsCl) in the presence of a base like potassium tert-butoxide, facilitating an intramolecular SN2 attack by the tertiary hydroxyl group. Alternatively, Mitsunobu conditions utilizing triphenylphosphine and diethyl azodicarboxylate (DEAD) can drive the cyclization under mild conditions. Acid-mediated cyclization using sulfuric acid is also viable for certain substrates. This mechanistic flexibility ensures that impurities can be managed effectively, as different cyclization conditions offer orthogonal purification opportunities, ultimately leading to high-purity 3-amino-oxetane derivatives suitable for sensitive biological applications.
How to Synthesize 3-Amino-Oxetane Derivatives Efficiently
The synthesis of these valuable heterocycles follows a logical progression from simple building blocks to the complex target structure, emphasizing operational simplicity and high yield. The process begins with the careful generation of the enolate from the glycine ester, followed by the addition of the ketone component to establish the core framework. Subsequent reduction converts the ester to an alcohol, setting the stage for the critical ring-closure event. The detailed standardized synthesis steps see the guide below for specific molar ratios, temperature profiles, and workup procedures optimized for reproducibility.
- Perform an aldol-type condensation between a glycine ester derivative (Compound II) and a ketone/aldehyde (Compound III) using a strong base like LDA or LiHMDS at low temperatures (-78°C to -5°C) to form the beta-hydroxy ester (Compound IV).
- Reduce the ester and hydroxyl functionalities of Compound IV using a hydride reducing agent such as lithium aluminum hydride (LiAlH4) or sodium borohydride (NaBH4) to generate the corresponding diol intermediate (Compound V).
- Execute an intramolecular cyclization of the diol Compound V using a cyclization reagent system, such as sulfuric acid, or a combination of base and sulfonyl chloride (TsCl/MsCl), or Mitsunobu conditions (PPh3/DEAD), to close the oxetane ring and yield Compound VI.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this synthesis route offers tangible benefits that extend beyond mere chemical elegance. The reliance on commodity chemicals such as acetone, glycine esters, and standard reducing agents means that raw material sourcing is robust and less susceptible to geopolitical disruptions or vendor monopolies. By avoiding specialized reagents like sodium azide, facilities can reduce the capital expenditure associated with safety infrastructure and hazardous waste disposal, leading to substantial cost savings in the overall production budget. Furthermore, the shortened synthetic sequence reduces the cumulative loss of material at each step, improving the overall mass balance and throughput of the manufacturing plant.
- Cost Reduction in Manufacturing: The elimination of hazardous azide reagents removes the need for expensive safety protocols and specialized waste treatment, directly lowering operational overhead. Additionally, the use of widely available bases and reducing agents ensures that reagent costs remain stable and predictable, avoiding the price volatility associated with exotic catalysts. The high efficiency of the cyclization step minimizes the need for extensive recycling of unreacted starting materials, further enhancing the economic viability of the process for large-scale production runs.
- Enhanced Supply Chain Reliability: Since the starting materials are bulk chemicals produced by multiple global suppliers, the risk of single-source dependency is significantly mitigated. This diversification of the supply base ensures that production schedules can be maintained even if one vendor faces disruptions. The robustness of the reaction conditions, which tolerate a range of solvents and temperatures, also means that manufacturing can be transferred between sites with minimal re-validation, providing flexibility in logistics and inventory management.
- Scalability and Environmental Compliance: The process is inherently scalable, having been demonstrated to work effectively from gram to kilogram scales without significant modification. The avoidance of heavy metal catalysts and toxic azides aligns with green chemistry principles, simplifying regulatory compliance and environmental permitting. Waste streams are primarily composed of organic salts and solvents that can be readily treated or recycled, reducing the environmental footprint and supporting sustainable manufacturing initiatives.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. These answers are derived directly from the experimental data and scope defined in the patent literature, providing a factual basis for decision-making.
Q: What are the primary advantages of this synthesis route over conventional azide displacement methods?
A: This novel route avoids the use of hazardous sodium azide and high-energy azide intermediates, significantly improving process safety. Additionally, it utilizes readily available glycine esters and ketones, shortening the synthetic sequence to just three steps compared to longer traditional pathways involving sulfonate activation and substitution.
Q: Can this method accommodate diverse substituents on the oxetane ring?
A: Yes, the method demonstrates high versatility. By varying Compound III (the carbonyl component), diverse substituents (R3 and R4) such as alkyl, phenyl, or spiro-cyclic groups can be introduced at the 3-position. Furthermore, the nitrogen protecting groups (R and R1) can be selected from benzyl, phthaloyl, or Boc-compatible precursors to suit downstream deprotection needs.
Q: Is this process suitable for large-scale commercial manufacturing?
A: The process is designed for scalability, utilizing standard industrial reagents like LDA, LiAlH4, and TsCl. The avoidance of exotic catalysts and the ability to purify intermediates via crystallization or standard column chromatography supports robust commercial scale-up from kilogram to multi-ton production levels.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 3-Amino-Oxetane Derivative Supplier
As the demand for complex heterocyclic building blocks continues to rise in the development of next-generation therapeutics, partnering with an experienced CDMO is crucial for success. NINGBO INNO PHARMCHEM possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project can transition smoothly from clinical trials to market launch. Our commitment to quality is underscored by our stringent purity specifications and rigorous QC labs, which utilize advanced analytical techniques to verify the identity and purity of every batch of 3-amino-oxetane derivatives we produce.
We invite you to engage with our technical procurement team to discuss your specific requirements for high-purity pharmaceutical intermediates. By requesting a Customized Cost-Saving Analysis, you can gain deeper insights into how our optimized synthesis routes can improve your project's economics. We encourage you to contact us today to obtain specific COA data and route feasibility assessments tailored to your unique molecular targets, ensuring a secure and efficient supply chain for your critical drug development programs.