Advanced Synthesis of High-Purity Para-Alkene Halobenzenes for Next-Generation Liquid Crystal Displays
Advanced Synthesis of High-Purity Para-Alkene Halobenzenes for Next-Generation Liquid Crystal Displays
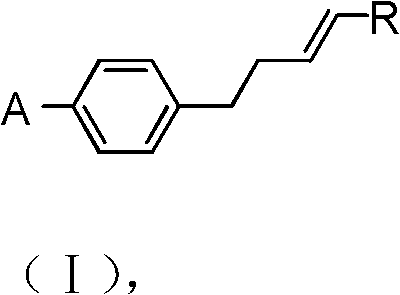
The rapid evolution of the display industry demands liquid crystal materials with increasingly precise electro-optical properties, necessitating intermediates of exceptional structural fidelity. Patent CN102126919B introduces a robust synthetic methodology for producing para-alkene chlorobenzene and para-alkene bromobenzene, which serve as critical precursors for liquid crystal monomers such as alkenyl benzoic ethers. These monomers are essential for modulating threshold voltage, enhancing response speed, and widening the operating temperature range of mixed liquid crystal formulations. The core innovation lies in a multi-step sequence that rigorously controls the position of the carbon-carbon double bond, effectively eliminating the formation of trace isomers that plague conventional synthesis routes. By integrating malonate alkylation, controlled decarboxylation, selective redox transformations, and a final Wittig olefination, this process delivers a high-quality intermediate suitable for high-end electronic applications.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditional synthetic pathways for alkenyl halobenzenes often rely on coupling reactions or less controlled elimination strategies that suffer from significant regioselectivity issues. A primary drawback identified in prior art, such as the technology disclosed in CN101555192, is the phenomenon of micro-double-bond migration during the build-up process. This migration results in the presence of trace isomeric impurities in the finished product, which can detrimentally affect the dielectric anisotropy and viscosity of the final liquid crystal mixture. Furthermore, conventional methods frequently exhibit unstable technology parameters, leading to fluctuating product yields and complicating the purification amplification process on an industrial scale. The inability to strictly dictate the double bond position means that extensive and costly purification steps are required to meet the stringent purity standards demanded by modern display manufacturers, thereby inflating production costs and extending lead times.
The Novel Approach
The methodology outlined in patent CN102126919B circumvents these historical challenges by employing a strategic sequence that anchors the double bond position in the final synthetic step. Instead of relying on coupling reactions where catalytic conditions might induce isomerization, this novel approach utilizes a Wittig reaction to construct the alkene moiety with absolute precision. This ensures that the double bond is located exactly where intended, preventing the formation of unwanted migratory isomers. Additionally, the process incorporates a highly efficient alkylation of dialkyl malonate followed by hydrolysis and decarboxylation, which builds the carbon skeleton with high atom economy. The subsequent reduction and oxidation steps utilize specific reagent systems, such as potassium borohydride with zinc chloride and TEMPO-mediated oxidation, to maintain functional group integrity throughout the synthesis, ultimately yielding a product with GC purity levels reaching 98% to 99%.
Mechanistic Insights into the Multi-Step Synthetic Route
The synthetic strategy begins with the alkylation of a para-halobenzyl halide (Formula IIa) with a dialkyl malonate (Formula IIb) under basic conditions at 50°C. This nucleophilic substitution extends the carbon chain by two atoms, establishing the propionic acid backbone required for the final structure. Following isolation, the diester intermediate undergoes hydrolysis and thermal decarboxylation at temperatures between 100°C and 120°C. This step is critical as it removes the ester groups and one carboxyl carbon, transforming the molecule into the corresponding 3-(4-halophenyl)propionic acid (Formula IV). The choice of base, typically sodium hydroxide or potassium hydroxide, and the controlled heating profile ensure complete conversion while minimizing side reactions that could degrade the aromatic ring or the halogen substituent.
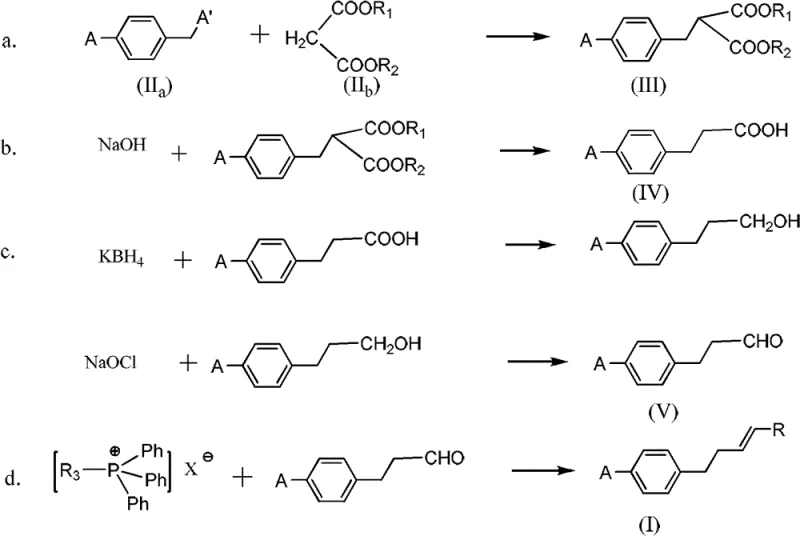
The transformation of the carboxylic acid to the requisite aldehyde (Formula V) involves a two-stage redox process designed for high selectivity. First, the acid is reduced to the primary alcohol using a borohydride system, specifically potassium borohydride activated by anhydrous zinc chloride in tetrahydrofuran. This activation allows for the reduction of the carboxylic acid under milder conditions than traditional lithium aluminum hydride, enhancing safety and handling. Subsequently, the alcohol is oxidized to the aldehyde using a TEMPO (2,2,6,6-tetramethylpiperidin-1-yl)oxyl and sodium hypochlorite system. This catalytic oxidation is highly chemoselective, stopping at the aldehyde stage without over-oxidizing to the carboxylic acid, which is vital for the success of the subsequent Wittig reaction. The final step involves reacting this aldehyde with a phosphonium salt (Formula VI) at low temperatures (-20°C to -15°C) to generate the target para-alkene halobenzene (Formula I) with defined stereochemistry and no double-bond migration.
How to Synthesize Para-Alkene Halobenzene Efficiently
The execution of this synthesis requires precise control over reaction parameters, particularly temperature and stoichiometry, to maximize yield and purity. The process is scalable and has been demonstrated to achieve an overall yield of approximately 62% with GC purity exceeding 98%. For R&D teams looking to implement this pathway, attention must be paid to the workup procedures, specifically the extraction and washing steps which remove inorganic salts and byproducts. The detailed standardized synthesis steps, including specific reagent quantities and isolation protocols for each intermediate, are provided in the guide below.
- Perform alkylation of para-halobenzyl halide with dialkyl malonate at 50°C to form the diester intermediate.
- Execute hydrolysis and decarboxylation at 100-120°C using sodium hydroxide to yield the corresponding phenylpropionic acid.
- Reduce the acid to alcohol using potassium borohydride and zinc chloride, followed by TEMPO-mediated oxidation to the aldehyde.
- Conduct a low-temperature Wittig reaction (-20 to -15°C) with a phosphonium salt to finalize the alkene structure without double-bond migration.
Commercial Advantages for Procurement and Supply Chain Teams
From a supply chain and procurement perspective, the adoption of this synthetic route offers substantial strategic benefits over legacy manufacturing processes. The elimination of double-bond migration impurities significantly reduces the burden on downstream purification units, which translates directly into lower processing costs and higher throughput. By achieving high purity at the synthesis stage, manufacturers can minimize the loss of valuable material during recrystallization or chromatography, thereby improving the overall mass balance of the production line. This efficiency is crucial for maintaining competitive pricing in the volatile market of electronic chemicals, where margin pressures are constant.
- Cost Reduction in Manufacturing: The process avoids the use of expensive transition metal catalysts often required for cross-coupling reactions, which not only lowers raw material costs but also eliminates the need for complex and costly heavy metal removal steps. The use of commodity reagents like dialkyl malonate, sodium hydroxide, and sodium hypochlorite ensures a stable and predictable cost structure. Furthermore, the high selectivity of the TEMPO oxidation and the Wittig reaction reduces the generation of difficult-to-separate byproducts, leading to significant savings in solvent usage and waste disposal fees associated with purification.
- Enhanced Supply Chain Reliability: The reliance on readily available starting materials such as para-halobenzyl halides and diethyl malonate mitigates the risk of supply disruptions common with specialized organometallic reagents. The robustness of the reaction conditions, particularly the tolerance of the alkylation and hydrolysis steps to minor variations, ensures consistent batch-to-batch quality. This reliability allows for more accurate production planning and inventory management, reducing the need for excessive safety stock and enabling a more responsive supply chain capable of meeting sudden spikes in demand from the display sector.
- Scalability and Environmental Compliance: The synthetic pathway is inherently scalable, having been validated from laboratory scale to multi-kilogram production without loss of efficiency. The avoidance of hazardous reagents and the ability to recycle solvents like tetrahydrofuran and dichloromethane align with increasingly stringent environmental regulations. The simplified workup procedures reduce the volume of aqueous waste generated, lowering the environmental footprint of the manufacturing process and facilitating easier compliance with local discharge standards, which is a critical factor for long-term operational continuity.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production of para-alkene halobenzenes using this patented methodology. These answers are derived directly from the experimental data and technical specifications provided in the patent documentation to ensure accuracy and relevance for potential partners.
Q: How does this synthesis method prevent double-bond migration?
A: Unlike coupling reactions that may cause isomerization, this method utilizes a Wittig reaction in the final step. The Wittig olefination allows for precise positioning of the double bond, ensuring the final product is free from trace isomers caused by migration.
Q: What is the expected purity of the final para-alkene halobenzene product?
A: The patent data indicates that the final product achieves a GC purity of 98% to 99%, which is significantly higher than the approximately 92% purity achievable with conventional linked reaction methods.
Q: Why is TEMPO/NaOCl used for the oxidation step?
A: The TEMPO/NaOCl system provides a mild and selective oxidation environment for converting the intermediate alcohol to the aldehyde. This selectivity minimizes side reactions and over-oxidation, preserving the integrity of the aromatic ring and the halogen substituent.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Para-Alkene Halobenzene Supplier
At NINGBO INNO PHARMCHEM, we recognize that the quality of liquid crystal materials is paramount to the performance of the final display panel. Our technical team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the intricate details of this four-step synthesis are managed with precision. We maintain stringent purity specifications and operate rigorous QC labs equipped with advanced GC-MS and HPLC systems to verify that every batch of para-alkene halobenzene meets the exacting standards required for high-end LCD applications. Our commitment to quality assurance guarantees that the intermediates we supply will perform consistently in your downstream monomer synthesis.
We invite you to collaborate with us to optimize your supply chain for liquid crystal intermediates. Our experts are ready to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements and purity needs. Please contact our technical procurement team today to request specific COA data and route feasibility assessments, and let us demonstrate how our advanced manufacturing capabilities can support your next-generation display projects.