Advanced Catalyst-Free Synthesis of N-Acyl Compounds for Commercial Pharmaceutical Manufacturing
Advanced Catalyst-Free Synthesis of N-Acyl Compounds for Commercial Pharmaceutical Manufacturing
The landscape of fine chemical manufacturing is constantly evolving towards safer, more efficient, and environmentally benign processes, particularly for high-value intermediates used in life sciences. Patent CN109824548B introduces a groundbreaking methodology for the preparation of N-acyl compounds, specifically targeting the synthesis of urea derivatives, carbamoyl fluorides, and heterocyclic structures like oxazolones and imidazolones. This technology leverages the unique reactivity of trifluoromethoxy-containing reagents, represented generally as R-OCF3, which possess the capability to decompose in situ to generate fluorophosgene. By bypassing the direct handling of hazardous phosgene gas and eliminating the need for transition metal catalysts, this approach offers a robust pathway for producing high-purity pharmaceutical intermediates. The reaction operates under remarkably mild conditions, typically ranging from -80°C to 100°C, with reaction times as short as 1 minute to 48 hours, demonstrating exceptional versatility for diverse amine substrates. For global procurement teams and R&D directors, this represents a significant opportunity to streamline supply chains for complex nitrogen-containing scaffolds while adhering to stricter environmental and safety regulations.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the industrial synthesis of urea derivatives and N-acyl compounds has been heavily reliant on the use of phosgene and its derivatives, or alternatively, the nucleophilic addition of isocyanates to amine compounds. While chemically effective, these traditional routes present severe logistical and safety challenges that impact the entire supply chain. Phosgene is a notorious chemical warfare agent with extreme toxicity, requiring specialized containment infrastructure, rigorous safety protocols, and expensive scrubbing systems to prevent accidental release. Furthermore, isocyanates, often used as safer alternatives to gaseous phosgene, are still highly reactive and toxic sensitizers that pose significant occupational health risks. In the context of carbamoyl fluoride synthesis, conventional methods often involve harsh fluorination conditions, such as electrochemical fluorination or halogen exchange reactions using corrosive reagents, which can lead to poor atom economy and difficult waste disposal issues. These factors collectively drive up the cost of goods sold (COGS) and extend lead times due to the complexity of regulatory compliance and safety auditing required for facilities handling such dangerous materials.
The Novel Approach
The methodology described in patent CN109824548B fundamentally shifts the paradigm by utilizing trifluoromethyl trifluoromethanesulfonate and its analogs (R-OCF3) as safe, solid, or liquid precursors that generate the active carbonylating species only within the reaction vessel. This in situ generation of fluorophosgene allows for the rapid N-carbonylation of amine substrates without the need to transport or store bulk toxic gases. The process is exceptionally broad in scope, accommodating primary amines to form symmetrical ureas, secondary amines to yield stable carbamoyl fluorides, and amino-alcohols to cyclize into valuable heterocycles. 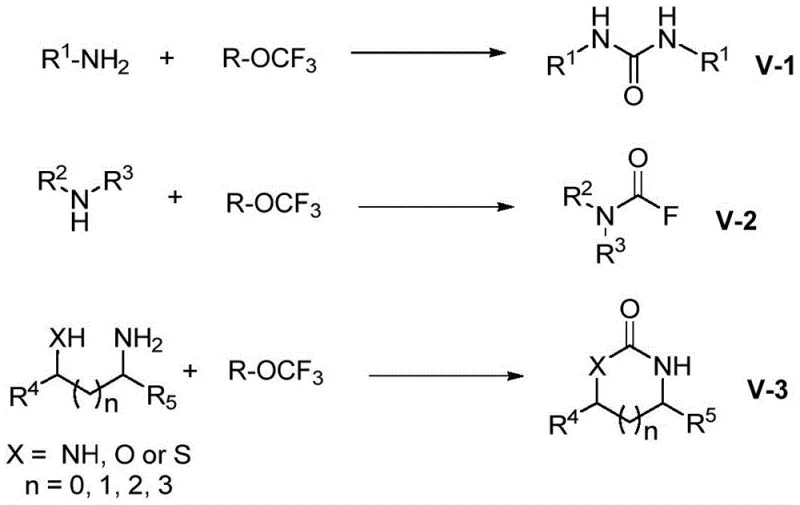 As illustrated in the reaction schemes, the transformation is highly selective and proceeds with high efficiency, often achieving yields exceeding 90% under optimized conditions. The absence of external catalysts or additives further simplifies the downstream processing, as there is no need for costly metal scavenging steps or complex filtration procedures to remove catalytic residues, making this an ideal candidate for cost reduction in pharmaceutical intermediate manufacturing.
As illustrated in the reaction schemes, the transformation is highly selective and proceeds with high efficiency, often achieving yields exceeding 90% under optimized conditions. The absence of external catalysts or additives further simplifies the downstream processing, as there is no need for costly metal scavenging steps or complex filtration procedures to remove catalytic residues, making this an ideal candidate for cost reduction in pharmaceutical intermediate manufacturing.
Mechanistic Insights into In-situ Fluorophosgene Generation
Understanding the mechanistic underpinnings of this transformation is critical for R&D directors aiming to adapt this chemistry for specific API intermediates. The core of this innovation lies in the instability of the trifluoromethoxy anion, which is generated upon the interaction of the R-OCF3 reagent with the amine substrate or fluoride anions present in the system. Once formed, this anion undergoes a rapid alpha-elimination process, decomposing to release fluorophosgene (COF2) and a fluoride ion. This fluorophosgene then acts as the electrophilic carbonyl source, reacting immediately with the nucleophilic amine to form a carbamoyl fluoride intermediate. 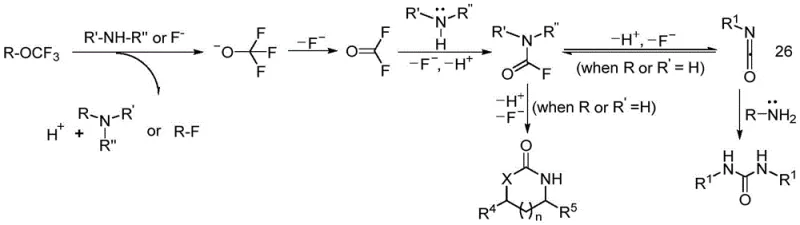 In the case of primary amines, this intermediate eliminates hydrogen fluoride to generate a highly reactive isocyanate species, which is subsequently trapped by a second equivalent of the amine to form the final urea derivative. For secondary amines, the reaction stops at the carbamoyl fluoride stage, providing access to this increasingly important pharmacophore without over-reaction. If the substrate contains a proximal hydroxyl or thiol group, intramolecular cyclization occurs preferentially, driven by entropy and the formation of stable five- or six-membered rings. This mechanistic clarity ensures that impurity profiles are predictable and manageable, as the reaction pathway avoids the formation of polymeric byproducts often seen in uncontrolled phosgenation reactions.
In the case of primary amines, this intermediate eliminates hydrogen fluoride to generate a highly reactive isocyanate species, which is subsequently trapped by a second equivalent of the amine to form the final urea derivative. For secondary amines, the reaction stops at the carbamoyl fluoride stage, providing access to this increasingly important pharmacophore without over-reaction. If the substrate contains a proximal hydroxyl or thiol group, intramolecular cyclization occurs preferentially, driven by entropy and the formation of stable five- or six-membered rings. This mechanistic clarity ensures that impurity profiles are predictable and manageable, as the reaction pathway avoids the formation of polymeric byproducts often seen in uncontrolled phosgenation reactions.
From an impurity control perspective, the mild nature of this reaction mechanism offers distinct advantages over traditional high-temperature or strong-base mediated processes. Because the active fluorophosgene is generated stoichiometrically and consumed rapidly within the solvent cage, the concentration of free toxic gas remains negligible, minimizing side reactions such as over-carbonylation or degradation of sensitive functional groups on the aromatic rings. The patent data indicates that electron-donating groups on aryl amines enhance reaction rates, while steric hindrance has a minimal impact, suggesting that even bulky, complex drug-like molecules can be processed effectively. Furthermore, the fluoride ion generated during the elimination step acts as a promoter for the decomposition of remaining trifluoromethyl esters, creating a self-accelerating cycle that ensures high conversion rates. This level of control is essential for maintaining the stringent purity specifications required for clinical-grade materials, reducing the burden on analytical quality control labs to identify and quantify trace toxic impurities associated with phosgene residues.
How to Synthesize N-Acyl Compounds Efficiently
Implementing this synthesis route in a laboratory or pilot plant setting requires careful attention to reagent stoichiometry and solvent selection to maximize yield and safety. The general protocol involves dissolving the amine substrate in a polar aprotic solvent such as acetonitrile, DMF, or dichloromethane, followed by the slow addition of the R-OCF3 reagent. The reaction can be conducted at temperatures ranging from cryogenic conditions (-80°C) to elevated temperatures (100°C), though room temperature or slightly below (-30°C to 30°C) is often preferred for balancing reaction rate and selectivity. Detailed standardized synthesis steps for specific substrates are provided in the guide below.
- Mix the amine compound (primary or secondary) with the trifluoromethoxy reagent (R-OCF3) in a suitable organic solvent such as acetonitrile or dichloromethane.
- Stir the reaction mixture at a temperature ranging from -80°C to 100°C, preferably between -30°C and 30°C, for a duration of 1 minute to 48 hours depending on substrate reactivity.
- Quench the reaction by adding water, remove the solvent under reduced pressure, and purify the resulting N-acyl compound via column chromatography or recrystallization.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this technology translates into tangible operational improvements beyond mere chemical yield. The elimination of phosgene from the supply chain removes a major bottleneck related to transportation regulations and storage licensing, allowing for more flexible manufacturing site selection. Additionally, the simplicity of the workup procedure—often requiring only water quenching and standard purification techniques like recrystallization or column chromatography—drastically reduces the consumption of solvents and energy compared to multi-step traditional syntheses. This streamlined workflow directly contributes to substantial cost savings in manufacturing overheads and waste treatment, aligning with modern green chemistry initiatives.
- Cost Reduction in Manufacturing: The most significant economic driver of this process is the complete absence of transition metal catalysts. In traditional cross-coupling or carbonylation reactions, the cost of palladium or nickel catalysts, combined with the expensive ligands and the subsequent need for metal scavengers to meet ppm-level specifications, can be prohibitive. By utilizing inexpensive trifluoromethoxy reagents that act as both reagent and activator, the raw material cost profile is significantly flattened. Furthermore, the high atom economy and the ability to use simple purification methods mean that less material is lost during isolation, improving the overall mass balance and reducing the cost per kilogram of the final API intermediate.
- Enhanced Supply Chain Reliability: Reliance on phosgene often ties production to a limited number of specialized facilities equipped to handle it, creating single points of failure in the supply chain. This new method utilizes reagents like trifluoromethyl trifluoromethanesulfonate which are more readily available and easier to transport globally. The robustness of the reaction across a wide temperature range (-80°C to 100°C) also means that manufacturing is less susceptible to disruptions caused by cooling system failures or heating limitations. This flexibility ensures consistent delivery schedules and reduces the risk of stockouts for critical intermediates, providing a more resilient supply base for downstream drug manufacturers.
- Scalability and Environmental Compliance: Scaling chemical processes that involve toxic gases is inherently risky and capital intensive. This catalyst-free protocol is ideally suited for scale-up because it avoids the engineering challenges associated with gas-liquid mixing and containment of hazardous vapors. The reaction generates minimal hazardous waste; the primary byproducts are fluoride salts and sulfonates, which are easier to treat than heavy metal sludge or chlorinated organic waste. This environmental profile simplifies the permitting process for new production lines and reduces long-term liability, making it a sustainable choice for large-scale commercial production of complex pharmaceutical intermediates.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this N-acyl compound synthesis technology. These answers are derived directly from the experimental data and technical specifications outlined in the patent documentation, ensuring accuracy for feasibility assessments.
Q: How does this method improve safety compared to traditional phosgene usage?
A: Traditional methods rely on toxic phosgene or isocyanates. This patent utilizes R-OCF3 reagents which generate fluorophosgene in situ only when needed, eliminating the need to store or handle bulk quantities of highly toxic gases, thereby significantly enhancing operational safety.
Q: What types of N-acyl products can be synthesized using this protocol?
A: The method is versatile: primary amines yield urea derivatives, secondary amines produce carbamoyl fluorides, and beta/gamma-amino alcohols undergo cyclization to form oxazolones, imidazolones, or thiazolones efficiently.
Q: Does this process require expensive transition metal catalysts?
A: No, the process is completely catalyst-free. It relies on the intrinsic reactivity of the trifluoromethoxy anion to decompose into fluorophosgene, which simplifies purification and reduces raw material costs associated with precious metal removal.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable N-Acyl Compound Supplier
As the demand for safer and more efficient synthetic routes grows, partnering with a CDMO that understands both the chemistry and the commercial implications is vital. NINGBO INNO PHARMCHEM possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that innovations like the R-OCF3 mediated carbonylation can be seamlessly transferred from the lab to the plant. Our facility is equipped with rigorous QC labs capable of verifying stringent purity specifications, ensuring that every batch of urea derivatives or carbamoyl fluorides meets the exacting standards required by global regulatory bodies. We understand that consistency is key in the pharmaceutical supply chain, and our robust process validation protocols guarantee batch-to-batch reproducibility.
We invite potential partners to engage with our technical team to explore how this technology can be applied to your specific pipeline. By requesting a Customized Cost-Saving Analysis, you can quantify the potential economic benefits of switching to this catalyst-free method for your specific targets. We encourage you to contact our technical procurement team to索取 specific COA data and route feasibility assessments, allowing you to make informed decisions based on real-world performance data rather than theoretical projections. Let us help you optimize your supply chain with cutting-edge chemistry that delivers both quality and value.