Advanced Synthesis of Midazolam Intermediates for Scalable Pharmaceutical Production
The pharmaceutical landscape for benzodiazepines continues to evolve, driven by the critical need for safer, more efficient manufacturing processes that comply with increasingly rigorous regulatory standards. Patent CN114773348A discloses a groundbreaking preparation method for Midazolam and its key intermediate M2, offering a robust alternative to legacy synthetic routes. Midazolam, chemically known as 1-methyl-8-chloro-6-(2-fluorophenyl)-4H-imidazo[1,5-a][1,4]benzodiazepine, is a vital active pharmaceutical ingredient (API) used globally for sedation, anxiolysis, and anesthesia induction. The structural complexity of this molecule, featuring a fused imidazobenzodiazepine core, has historically presented significant challenges in process chemistry, particularly regarding impurity control and operational safety.
This technical insight report analyzes the novel methodology presented in the patent, which utilizes a strategic protection-deprotection sequence centered around intermediate M2. By shifting away from hazardous nitrosation and hydrogenation steps, this route addresses critical pain points for reliable API intermediate suppliers and manufacturing partners. The process begins with the protection of the starting material SM1, followed by a highly selective reductive amination, and concludes with a streamlined cyclization and oxidation sequence. This approach not only shortens the overall synthetic timeline but also drastically reduces the generation of genotoxic impurities, a paramount concern for modern drug substance production.
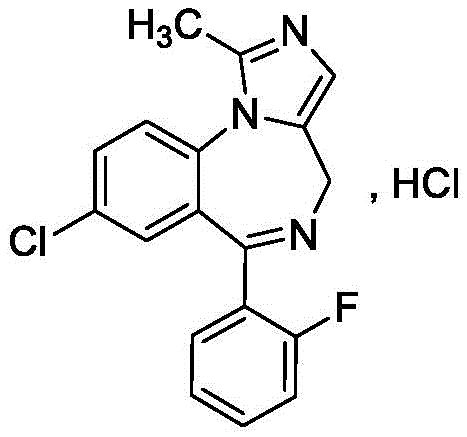
The limitations of conventional methods for synthesizing Midazolam are well-documented in chemical literature and pose substantial barriers to efficient cost reduction in pharmaceutical manufacturing. Traditional Route 1, often cited in prior art, relies on an eight-step sequence that begins with the acylation of SM1 using chloroacetyl chloride. This reagent is highly toxic and classified as a controlled substance in many jurisdictions, creating severe supply chain bottlenecks and transportation hazards. Furthermore, the route necessitates the use of methylamine gas or solutions, which are also controlled reagents, complicating procurement and increasing operational overhead for production facilities.
Beyond reagent toxicity, the conventional pathway involves eighteen key monitoring reactions with elevated safety risks, including multiple halogenation and ammoniation steps. A critical flaw in these legacy methods is the reliance on excessive amounts of Raney nickel catalyst for hydrogenation. Raney nickel is pyrophoric and poses significant explosion risks during scale-up, requiring specialized equipment and rigorous safety protocols that drive up capital expenditure. Additionally, the use of sodium nitrite in the nitrosation step inevitably leads to the formation of nitrosamine impurities. These genotoxic contaminants are strictly regulated due to their carcinogenic potential, necessitating expensive and complex purification strategies to ensure patient safety. The cumulative effect of these factors results in a low total yield of approximately 6.7%, rendering the process economically inefficient for large-scale operations.
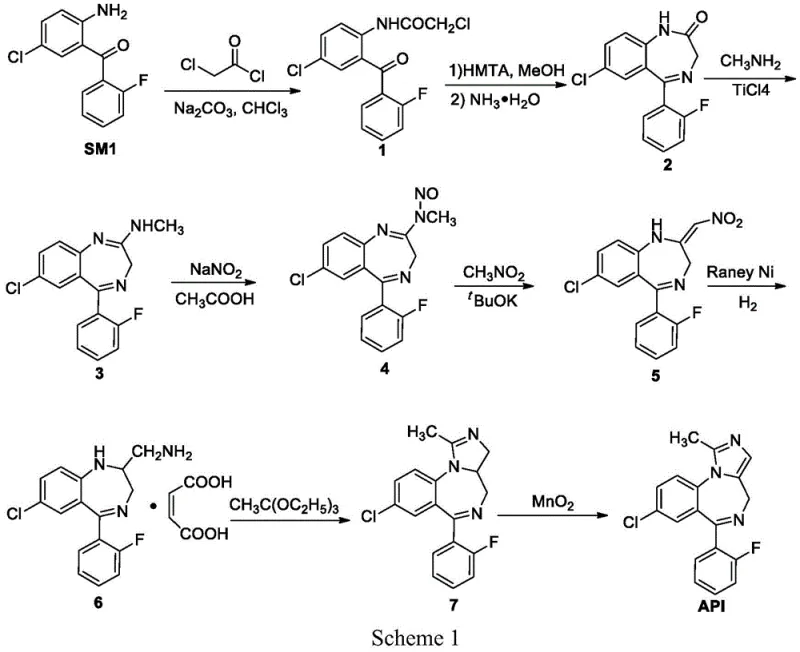
In stark contrast, the novel approach detailed in the patent introduces a paradigm shift by utilizing intermediate M2 as a pivotal junction in the synthesis. This strategy bypasses the dangerous nitrosation and high-risk hydrogenation steps entirely. The core innovation lies in the initial protection of the ketone functionality in SM1 using alcohols or thiols to form intermediate M1. This protection group serves a dual purpose: it prevents unwanted side reactions at the carbonyl center during subsequent amination and facilitates the construction of the seven-membered diazepine ring later in the sequence. The subsequent reaction of M1 with SM2, a protected 1,3-diaminoacetone derivative, proceeds via a clean reductive amination mechanism.
The novelty of this route is further exemplified by its flexibility in reagent selection. The patent demonstrates that various protecting groups, such as ethylene glycol or ethanedithiol, can be employed effectively. For instance, using ethanedithiol with titanium tetrachloride yields the thioketal intermediate M1 with high efficiency. Alternatively, ethylene glycol with trimethylchlorosilane offers a metal-free protection strategy. This adaptability allows manufacturers to optimize the process based on available raw materials and waste treatment capabilities. The elimination of genotoxic nitrosamines and pyrophoric catalysts fundamentally alters the safety profile of the production line, making it inherently safer for operators and more compatible with standard pharmaceutical manufacturing equipment. This translates directly into commercial scale-up of complex pharmaceutical intermediates with reduced regulatory friction.
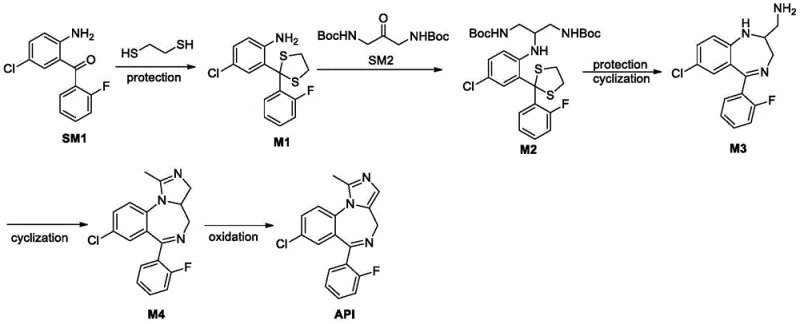
The mechanistic insights into this catalytic sequence reveal a sophisticated orchestration of organic transformations designed to maximize selectivity and minimize byproduct formation. The formation of intermediate M2 via reductive amination is the linchpin of this synthesis. In this step, the amine group of the protected benzophenone (M1) condenses with the ketone of the protected diaminoacetone (SM2) to form an imine intermediate. This imine is then selectively reduced using mild hydride sources such as sodium cyanoborohydride or sodium triacetoxyborohydride. The choice of reducing agent is critical; unlike harsh catalytic hydrogenation, these chemical reducers operate under mild acidic or neutral conditions, preserving the sensitive protecting groups and preventing the reduction of other functional moieties like the aryl chloride or fluoride.
Impurity control is rigorously maintained throughout the mechanism. By avoiding sodium nitrite, the pathway eliminates the risk of N-nitrosation, a common failure mode in benzodiazepine synthesis that leads to hard-to-remove genotoxic alerts. Furthermore, the cyclization step utilizes trimethyl orthoacetate to construct the imidazole ring. This reaction proceeds through an acid-catalyzed condensation that is highly atom-economical. The final oxidation step, employing manganese dioxide in dimethyl sulfoxide, aromatizes the imidazole ring to yield the final API. This oxidation method is superior to traditional methods as it avoids over-oxidation and chlorination side reactions. The result is a product with exceptional purity, reported in the patent examples to reach 99.92% by HPLC, demonstrating the robustness of the mechanistic design in suppressing trace impurities.
How to Synthesize Midazolam Efficiently
The synthesis of Midazolam via the M2 intermediate represents a significant optimization over traditional methods, offering a clear pathway for process chemists to implement in pilot and production plants. The procedure leverages widely available starting materials and avoids the logistical nightmares associated with controlled reagents. The initial protection step can be conducted in common solvents like dichloromethane or toluene, and the subsequent reductive amination tolerates a range of conditions, providing flexibility for process optimization. The final cyclization and oxidation steps are high-yielding and produce minimal waste, aligning with green chemistry principles. For detailed operational parameters, stoichiometry, and workup procedures, please refer to the standardized synthesis guide below.
- Protect the ketone group of SM1 (2-amino-5-chloro-2'-fluorobenzophenone) using ethanedithiol or ethylene glycol to form intermediate M1.
- Perform reductive amination between intermediate M1 and protected 1,3-diaminoacetone (SM2) using a reducing agent like sodium cyanoborohydride to obtain intermediate M2.
- Deprotect M2, cyclize with trimethyl orthoacetate to form the imidazole ring (M4), and oxidize with MnO2/DMSO to yield final Midazolam.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this novel synthesis route offers transformative advantages that extend beyond simple yield improvements. The primary value driver is the substantial reduction in operational risk and associated costs. By eliminating the need for chloroacetyl chloride and methylamine, companies can bypass the stringent licensing, storage, and transportation requirements mandated for these controlled substances. This simplification of the raw material portfolio significantly lowers the administrative burden and insurance costs associated with hazardous material handling. Furthermore, the removal of Raney nickel from the process eliminates the need for specialized pyrophoric handling equipment and the complex disposal protocols required for spent nickel catalysts, leading to direct savings in waste management and facility maintenance.
Enhanced supply chain reliability is another critical benefit derived from this technology. The reliance on commodity chemicals like ethylene glycol, ethanedithiol, and standard reducing agents ensures a stable and diversified supply base, reducing vulnerability to single-source supplier disruptions. The shorter synthetic route, characterized by fewer unit operations and simplified purification steps, inherently reduces the production lead time. This agility allows manufacturers to respond more rapidly to market demand fluctuations, ensuring consistent availability of this critical sedative API. Additionally, the high purity of the crude product minimizes the need for extensive recrystallization or chromatographic purification, thereby increasing throughput and reducing solvent consumption. These efficiencies collectively contribute to a more resilient and cost-effective supply chain architecture.
Scalability and environmental compliance are seamlessly integrated into this process design. The mild reaction conditions, typically ranging from ambient temperature to moderate heating (e.g., 110°C for cyclization), are easily manageable in standard glass-lined or stainless steel reactors, facilitating seamless technology transfer from laboratory to commercial scale. The avoidance of genotoxic reagents simplifies the cleaning validation processes between batches, reducing downtime and cross-contamination risks. From an environmental perspective, the reduction in hazardous waste streams and the use of less toxic reagents align with global sustainability goals and increasingly strict environmental regulations. This proactive approach to environmental stewardship not only mitigates regulatory risk but also enhances the corporate social responsibility profile of the manufacturing entity, making it a preferred partner for global pharmaceutical clients.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this novel Midazolam synthesis route. These answers are derived directly from the experimental data and technical disclosures within the patent documentation, providing a factual basis for decision-making. Understanding these nuances is essential for R&D teams evaluating process feasibility and procurement teams assessing supplier capabilities. The focus is on clarity regarding safety, purity, and scalability to ensure all stakeholders have a comprehensive understanding of the technology's value proposition.
Q: How does this new synthesis route address genotoxic impurity concerns?
A: Unlike traditional routes that utilize sodium nitrite and generate nitrosamine impurities, this novel method avoids nitrosation reactions entirely, significantly enhancing product safety and regulatory compliance.
Q: What are the scalability advantages of using intermediate M2?
A: The process eliminates the use of hazardous reagents like chloroacetyl chloride and pyrophoric Raney nickel, replacing them with milder conditions and safer catalysts, which simplifies equipment requirements and allows for safer large-scale production.
Q: What purity levels can be achieved with this method?
A: The patent data indicates that the final Midazolam product can achieve HPLC purity levels exceeding 99.9%, meeting stringent pharmaceutical specifications without complex purification steps.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Midazolam Supplier
At NINGBO INNO PHARMCHEM, we recognize that the transition from patent concept to commercial reality requires a partner with deep technical expertise and robust manufacturing capabilities. As a leading CDMO, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the theoretical benefits of this novel route are fully realized in practice. Our state-of-the-art facilities are equipped to handle the specific reagents and conditions required for the M2 intermediate synthesis, including corrosion-resistant reactors for acid-catalyzed steps and advanced filtration systems for high-purity isolation. We maintain stringent purity specifications and operate rigorous QC labs to guarantee that every batch of Midazolam meets or exceeds international pharmacopoeia standards.
We invite you to collaborate with us to leverage this advanced synthesis technology for your supply chain. Our technical team is prepared to conduct a Customized Cost-Saving Analysis tailored to your specific volume requirements and quality targets. By partnering with NINGBO INNO PHARMCHEM, you gain access to a secure, compliant, and efficient source of high-quality Midazolam. We encourage you to contact our technical procurement team today to request specific COA data and route feasibility assessments, and let us demonstrate how our engineering excellence can drive value for your organization.