Advanced Manufacturing of 7,8-Dimethoxy-1,3-Dihydro-2H-3-Benzazepin-2-One for Global Pharma Supply Chains
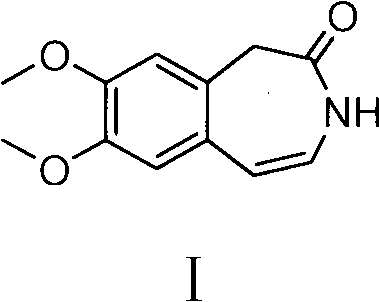
The pharmaceutical industry continuously seeks robust synthetic routes for critical cardiovascular drug intermediates, and Patent CN102276530B presents a transformative approach to manufacturing 7,8-dimethoxy-1,3-dihydro-2H-3-benzazepin-2-one. This compound serves as the pivotal scaffold for Ivabradine, a widely prescribed agent for treating angina pectoris and chronic heart failure. The disclosed methodology fundamentally shifts the synthetic paradigm from hazardous acid chloride chemistry to a milder, more efficient active ester activation strategy. By replacing thionyl chloride with coupling agents such as 2,4-dimethoxy-6-chloro-1,3,5-s-triazine, the process mitigates severe equipment corrosion and environmental pollution risks associated with hydrogen chloride gas evolution. This technical breakthrough not only enhances the safety profile of the manufacturing facility but also delivers a substantial improvement in overall reaction yield, pushing efficiency metrics from historical lows of 58 percent to a robust range of 75 to 80 percent. For global procurement teams, this represents a vital opportunity to secure a reliable pharmaceutical intermediates supplier capable of delivering high-quality materials with reduced supply chain volatility.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of this benzazepinone derivative relied heavily on the formation of an acid chloride intermediate using thionyl chloride, a reagent notorious for its aggressive reactivity and hazardous byproducts. In these legacy processes, the reaction of 3,4-dimethoxyphenylacetic acid with thionyl chloride generates copious amounts of hydrogen chloride gas, which imposes severe corrosive stress on reactor vessels and scrubbing systems, leading to frequent maintenance downtime and increased capital expenditure. Furthermore, the resulting acid chloride is thermally unstable and requires rigorous purification via high-temperature vacuum distillation to remove residual thionyl chloride and byproducts before it can be safely reacted with the amine component. This energy-intensive distillation step not only escalates operational costs but also introduces significant thermal degradation risks that compromise the purity of the intermediate. Consequently, the cumulative yield of these multi-step conventional routes often stagnates around 58 percent, creating a bottleneck for cost reduction in API manufacturing and limiting the ability to meet surging market demand efficiently.
The Novel Approach
In stark contrast, the novel methodology detailed in the patent utilizes an in situ activation strategy that bypasses the isolation of unstable acid chlorides entirely. By activating the carboxyl group of the starting acid directly with coupling reagents like 2,4-dimethoxy-6-chloro-1,3,5-s-triazine or DCC, the process forms a highly reactive active ester species under mild conditions. This active ester can then undergo immediate amidation with 2,2-dimethoxyethylamine in the same reaction vessel, effectively telescoping two synthetic steps into a seamless operation. This one-pot capability drastically reduces solvent consumption, minimizes material transfer losses, and eliminates the need for the aforementioned high-temperature distillation. The subsequent cyclization step proceeds smoothly under acidic conditions to close the seven-membered ring, delivering the target molecule with exceptional purity. This streamlined workflow exemplifies the commercial scale-up of complex pharmaceutical intermediates, offering a pathway that is not only chemically superior but also economically optimized for modern green chemistry standards.
Mechanistic Insights into Active Ester-Mediated Cyclization
The core innovation of this synthesis lies in the precise control of nucleophilic acyl substitution through active ester intermediates. When 3,4-dimethoxyphenylacetic acid is treated with a coupling agent in the presence of a tertiary amine base such as N-methylmorpholine, the carboxylate anion attacks the electrophilic center of the activator. This forms an O-acylisourea or a triazine-based active ester, which possesses significantly higher electrophilicity than the parent carboxylic acid but greater stability than an acid chloride. This enhanced stability allows the intermediate to persist in the reaction mixture at temperatures ranging from -20°C to 35°C without decomposing, providing a wide operational window for the addition of the amine nucleophile. The amine then attacks the carbonyl carbon of the active ester, displacing the leaving group and forming the amide bond with high fidelity. This mechanism effectively suppresses side reactions such as racemization or over-acylation, which are common pitfalls in aggressive acid chloride chemistry, thereby ensuring the structural integrity of the sensitive dimethoxy-substituted aromatic ring.
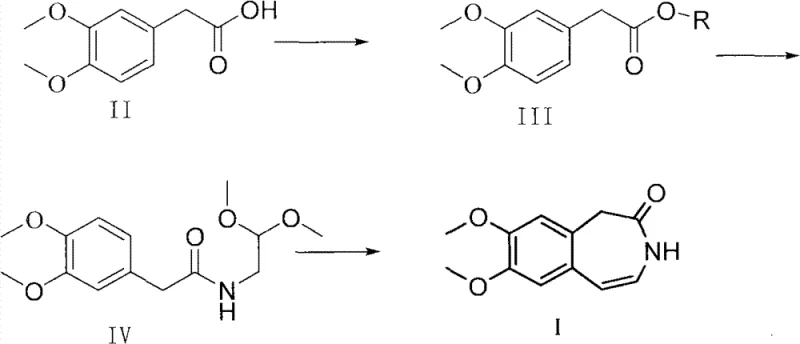
Following the formation of the linear amide precursor, the final cyclization step involves an intramolecular electrophilic aromatic substitution or a condensation reaction facilitated by strong acid catalysis. The use of concentrated hydrochloric acid combined with glacial acetic acid protonates the acetal or imine functionality within the side chain, generating a highly reactive iminium ion species. This electrophile is then attacked by the electron-rich aromatic ring at the ortho position relative to the methoxy groups, closing the seven-membered benzazepine ring. The mild nature of the preceding activation steps ensures that the starting material entering this cyclization phase is of high purity, which is critical for minimizing the formation of polymeric byproducts or regioisomers during ring closure. By controlling the acidity and temperature during this final stage, manufacturers can achieve high-purity benzazepinone specifications that meet stringent regulatory requirements for downstream drug substance synthesis, ultimately reducing lead time for high-purity intermediates required for clinical and commercial batches.
How to Synthesize 7,8-Dimethoxy-1,3-Dihydro-2H-3-Benzazepin-2-One Efficiently
Executing this synthesis requires careful attention to temperature control and reagent stoichiometry to maximize the benefits of the active ester pathway. The process begins with the dissolution of the activator in a suitable solvent like dichloromethane, followed by the addition of the carboxylic acid substrate at cooled temperatures to manage the exotherm of activation. Once the active ester is formed, the amine component is introduced slowly to maintain reaction homogeneity and prevent localized overheating. After the amidation is complete, the reaction mixture can be worked up to isolate the amide intermediate or proceeded directly to cyclization depending on the specific facility capabilities. The detailed standardized operating procedures, including specific molar ratios, stirring rates, and quenching protocols necessary for reproducible GMP manufacturing, are outlined in the technical guide below.
- Activate 3,4-dimethoxyphenylacetic acid using 2,4-dimethoxy-6-chloro-1,3,5-s-triazine and a base like N-methylmorpholine in dichloromethane at low temperatures.
- React the resulting active ester intermediate directly with 2,2-dimethoxyethylamine to form the amide precursor without isolation.
- Perform acid-catalyzed cyclization using concentrated hydrochloric acid and glacial acetic acid, followed by precipitation in ice water to isolate the final product.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this patented synthetic route offers profound strategic advantages beyond simple yield improvements. By eliminating the reliance on thionyl chloride, facilities can significantly reduce the costs associated with hazardous waste disposal and corrosion-resistant infrastructure maintenance. The removal of high-temperature vacuum distillation steps translates directly into lower energy consumption and reduced cycle times, allowing for faster throughput and improved asset utilization. Furthermore, the use of commercially available and stable coupling reagents ensures a resilient supply chain that is less susceptible to the logistical disruptions often associated with hazardous gas transport. This operational stability is crucial for maintaining continuous production schedules and meeting just-in-time delivery commitments to downstream pharmaceutical partners.
- Cost Reduction in Manufacturing: The elimination of thionyl chloride removes the need for specialized scrubbing systems and corrosion-resistant alloys in reactor construction, leading to substantial capital expenditure savings. Additionally, the avoidance of energy-intensive distillation steps reduces utility costs, while the higher overall yield means less raw material is required per kilogram of finished product, driving down the cost of goods sold significantly.
- Enhanced Supply Chain Reliability: Sourcing stable solid activators like triazine derivatives is logistically simpler and safer than managing bulk shipments of corrosive liquids like thionyl chloride. This simplification reduces regulatory burdens and transportation risks, ensuring a more consistent flow of raw materials into the production line and minimizing the risk of shutdowns due to supply shortages or safety incidents.
- Scalability and Environmental Compliance: The process generates significantly less acidic waste gas, simplifying effluent treatment and ensuring compliance with increasingly strict environmental regulations. The milder reaction conditions and simplified workup procedures make the technology highly scalable from pilot plant to multi-ton commercial production without the engineering complexities associated with handling large volumes of toxic gases.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this advanced synthesis route. These answers are derived directly from the experimental data and process descriptions found in the patent literature, providing a factual basis for evaluating the technology's fit within your existing manufacturing portfolio. Understanding these nuances is essential for making informed decisions about process validation and vendor qualification.
Q: Why is the active ester method superior to the traditional thionyl chloride route?
A: The active ester method avoids the generation of corrosive hydrogen chloride gas and eliminates the need for energy-intensive high-temperature vacuum distillation, resulting in significantly higher yields and reduced equipment maintenance costs.
Q: What are the critical reaction conditions for maximizing yield in this synthesis?
A: Maintaining the activation temperature between -10°C and 20°C is crucial to prevent side reactions. Additionally, using activators like 2,4-dimethoxy-6-chloro-1,3,5-s-triazine ensures high reactivity without requiring harsh purification steps.
Q: Is this process suitable for large-scale industrial production?
A: Yes, the process is specifically designed for industrial scalability by removing hazardous reagents like thionyl chloride and simplifying the workup procedure, which facilitates safer handling and consistent batch-to-batch quality.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 7,8-Dimethoxy-1,3-Dihydro-2H-3-Benzazepin-2-One Supplier
At NINGBO INNO PHARMCHEM, we recognize that the transition to greener, more efficient synthetic routes is critical for the long-term sustainability of the pharmaceutical supply chain. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the theoretical benefits of this active ester chemistry are fully realized in a GMP-compliant environment. We operate stringent purity specifications and maintain rigorous QC labs to guarantee that every batch of 7,8-dimethoxy-1,3-dihydro-2H-3-benzazepin-2-one meets the exacting standards required for Ivabradine synthesis. Our commitment to technical excellence allows us to offer a level of quality assurance that supports your regulatory filings and accelerates your time to market.
We invite you to engage with our technical procurement team to discuss how this optimized route can enhance your specific project economics. By requesting a Customized Cost-Saving Analysis, you can gain detailed insights into the potential ROI of switching to this methodology. We encourage you to contact us today to obtain specific COA data and route feasibility assessments tailored to your volume requirements, ensuring a partnership built on transparency, scientific rigor, and mutual growth.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →