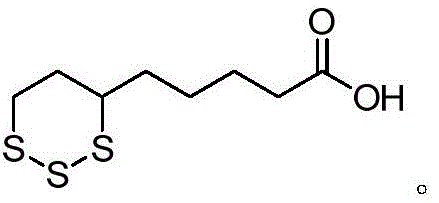
Advanced Synthesis of Lipoic Acid Impurity A for Pharmaceutical Quality Control
The pharmaceutical industry demands rigorous quality control standards, particularly for active pharmaceutical ingredients (APIs) like lipoic acid, where impurity profiling is critical for regulatory compliance. Patent CN113234059A introduces a significant technological advancement in the preparation of Lipoic Acid Impurity A, also known chemically as 1,2,3-Trithiane-4-pentanoic acid. This specific impurity serves as an essential reference standard for detecting degradation products in lipoic acid bulk drugs and formulations. The disclosed method overcomes historical challenges associated with harsh reaction conditions and complex purification steps, offering a streamlined three-step synthesis that operates under mild temperatures. By utilizing a strategic sequence of oxidation, hydrolytic ring-opening, and cyclization, this process ensures the production of high-purity material suitable for analytical applications.
Traditional methods for synthesizing trithiane derivatives often involve severe thermal conditions and labor-intensive purification techniques that hinder efficient production. Prior art, such as the method disclosed in CN107652264A, typically requires high-temperature reflux reactions and relies heavily on silica gel column chromatography for isolation. These conventional approaches not only increase operational complexity and time but also introduce potential safety hazards due to the handling of hot reactive mixtures and large volumes of solvents required for chromatography. Furthermore, the reliance on column chromatography creates a bottleneck for scalability, making it difficult to transition from laboratory benchtop synthesis to industrial manufacturing without incurring prohibitive costs and yield losses.
In stark contrast, the novel approach detailed in the patent utilizes a温和 (mild) reaction pathway that drastically simplifies the workflow. The new method replaces high-temperature reflux with controlled low-temperature oxidation and ambient temperature hydrolysis. Crucially, it substitutes the cumbersome column chromatography step with a robust crystallization protocol using mixed solvents. This shift not only enhances the safety profile of the manufacturing process by reducing thermal risks but also significantly improves the throughput capability. The ability to isolate the target compound through crystallization rather than chromatography represents a major leap forward in process chemistry, aligning perfectly with the principles of green chemistry and efficient resource utilization.
Mechanistic Insights into Oxidative Ring-Opening and Cyclization
The core of this synthetic strategy lies in the precise manipulation of sulfur oxidation states and ring dynamics. The process initiates with the selective oxidation of the disulfide bond in lipoic acid using m-chloroperoxybenzoic acid (mCPBA) in a chloroform solvent system. This step converts the stable five-membered dithiolane ring into a sulfoxide intermediate (Compound B), activating the molecule for subsequent nucleophilic attack. The reaction is carefully maintained at 0-10°C to prevent over-oxidation to the sulfone, ensuring high selectivity for the desired sulfoxide species which is pivotal for the next transformation.
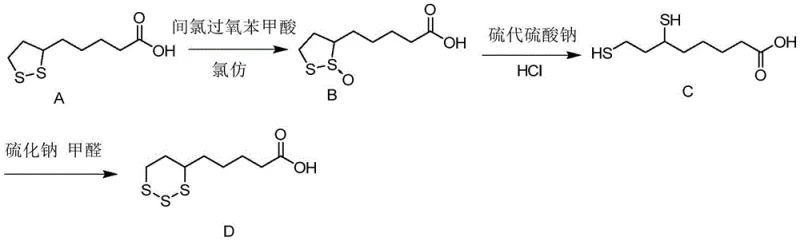
Following oxidation, the sulfoxide intermediate undergoes a hydrolytic ring-opening reaction mediated by sodium thiosulfate and hydrochloric acid. This step effectively cleaves the sulfur-sulfur bonds, generating a linear dithiol species (Compound C) while maintaining the carbon backbone integrity. The use of a phase transfer catalyst, such as tetrabutylammonium bromide, facilitates the interaction between the organic substrate and the aqueous inorganic reagents, enhancing reaction efficiency. Finally, the linear dithiol is subjected to a cyclization reaction with sodium sulfide and formaldehyde. This condensation reaction reconstructs a six-membered trithiane ring, yielding the final Lipoic Acid Impurity A. The mechanistic control at each stage minimizes the formation of side products, directly contributing to the high purity observed in the final isolate.
How to Synthesize Lipoic Acid Impurity A Efficiently
The synthesis of Lipoic Acid Impurity A via this patented route involves a logical progression of functional group transformations that are amenable to standard chemical processing equipment. The procedure begins with the dissolution of the starting material in an appropriate organic solvent, followed by the controlled addition of the oxidant. Subsequent steps involve aqueous workups and extractions, which are standard unit operations in any chemical plant. The final purification is achieved through a temperature-controlled crystallization process, which is far more scalable than chromatographic methods. For detailed operational parameters, stoichiometry, and specific workup procedures, please refer to the standardized guide below.
- Oxidize lipoic acid with m-chloroperoxybenzoic acid in chloroform at 0-10°C to obtain the sulfoxide intermediate (Compound B).
- Perform hydrolytic ring-opening of Compound B using sodium thiosulfate and hydrochloric acid with a phase transfer catalyst to yield the dithiol intermediate (Compound C).
- React Compound C with sodium sulfide and formaldehyde in tetrahydrofuran, followed by crystallization to isolate pure Lipoic Acid Impurity A.
Commercial Advantages for Procurement and Supply Chain Teams
From a commercial perspective, this synthesis route offers substantial benefits for procurement managers and supply chain directors looking to optimize their sourcing strategies for pharmaceutical intermediates. The elimination of high-temperature reflux and column chromatography translates directly into reduced energy consumption and lower solvent usage. By avoiding the need for large quantities of silica gel and eluents, the process significantly lowers the variable costs associated with production. Furthermore, the simplified workflow reduces the manpower hours required for execution, allowing for faster batch turnover times and improved overall equipment effectiveness (OEE).
- Cost Reduction in Manufacturing: The removal of column chromatography is a primary driver for cost savings. Chromatography is inherently expensive due to the cost of stationary phases and the large volumes of solvents required for elution, which subsequently need to be recovered or disposed of. By replacing this with crystallization, the process achieves a drastic reduction in material costs and waste treatment expenses. Additionally, the use of common, commercially available reagents like sodium thiosulfate and formaldehyde ensures that raw material costs remain stable and predictable, avoiding the volatility associated with specialized catalysts.
- Enhanced Supply Chain Reliability: The mild reaction conditions (0-45°C) reduce the stress on reactor vessels and ancillary equipment, leading to lower maintenance requirements and higher asset availability. The process does not require exotic or hard-to-source reagents; all key inputs are commodity chemicals with robust global supply chains. This universality of raw materials mitigates the risk of supply disruptions, ensuring a continuous and reliable flow of the intermediate to downstream users. The stability of the process also means fewer failed batches, further securing the supply line.
- Scalability and Environmental Compliance: Scalability is inherently built into this design due to the reliance on liquid-phase reactions and crystallization, which are easily transferred from pilot plants to multi-ton reactors. The process generates less hazardous waste compared to traditional methods, as it avoids the disposal of spent silica gel and complex solvent mixtures typical of chromatography. This alignment with environmental regulations simplifies the permitting process for manufacturing facilities and reduces the long-term liability associated with waste management, making it a sustainable choice for long-term production.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of Lipoic Acid Impurity A. These answers are derived directly from the technical specifications and beneficial effects outlined in the patent documentation, providing clarity on the method's capabilities and limitations. Understanding these details is crucial for R&D teams evaluating the material for method validation and for procurement teams assessing supplier qualifications.
Q: What are the key advantages of this new synthesis method for Lipoic Acid Impurity A?
A: The method described in patent CN113234059A utilizes mild reaction conditions (0-45°C) compared to traditional high-temperature reflux methods. It eliminates the need for complex column chromatography, relying instead on crystallization for purification, which significantly simplifies the operation and improves scalability.
Q: What is the expected purity of the final product using this route?
A: The patent data indicates that the final product, Lipoic Acid Impurity A, can achieve a purity of approximately 99.7% as detected by HPLC. This high level of purity is critical for its use as a reference standard in pharmaceutical quality control.
Q: Is this process suitable for large-scale manufacturing?
A: Yes, the process is highly suitable for scale-up. The avoidance of hazardous high-temperature steps and the replacement of silica gel chromatography with standard extraction and crystallization techniques make it economically viable and safer for commercial production environments.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Lipoic Acid Impurity A Supplier
NINGBO INNO PHARMCHEM stands at the forefront of fine chemical manufacturing, leveraging advanced synthetic methodologies like the one described in CN113234059A to deliver superior pharmaceutical intermediates. Our facility is equipped with state-of-the-art reactors capable of handling diverse reaction types, from cryogenic oxidations to exothermic cyclizations, ensuring that complex pathways are executed with precision. We possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, guaranteeing that our clients receive consistent quality regardless of order volume. Our commitment to excellence is underpinned by stringent purity specifications and rigorous QC labs that utilize HPLC and NMR to verify every batch against the highest industry standards.
We invite global partners to collaborate with us to secure a stable supply of high-quality Lipoic Acid Impurity A. Our technical team is ready to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements, demonstrating how our optimized processes can reduce your total cost of ownership. We encourage you to contact our technical procurement team today to request specific COA data and route feasibility assessments, ensuring that your quality control protocols are supported by the most reliable reference materials available in the market.