Scalable Synthesis of N-Substituted Benzamide Derivatives for Oncology Applications
Introduction to Advanced Kinase Inhibitor Synthesis
The pharmaceutical landscape is continuously evolving with the demand for potent dual-action therapeutics, specifically those targeting both protein kinases and histone deacetylases (HDACs). Patent CN102775389A introduces a sophisticated yet practical methodology for preparing N-substituted-4-(7-chloroquinoline-4-amino)-benzamide derivatives, compounds that have demonstrated significant potential in treating oncology and cardiovascular conditions. This technical disclosure outlines a streamlined three-step synthetic pathway that begins with readily available starting materials like p-aminobenzoic acid and 4,7-dichloroquinoline. For R&D directors and procurement specialists, understanding the nuances of this route is critical, as it offers a balance between chemical complexity and manufacturability. The resulting molecules exhibit robust inhibitory activity against various cancer cell lines, including lung and liver carcinoma, making them high-value targets for API development. By leveraging this patented approach, manufacturers can access a reliable supply chain for complex heterocyclic intermediates essential for next-generation cancer therapies.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditionally, the synthesis of complex quinoline-pyrimidine hybrids often involves cumbersome multi-step sequences that suffer from low overall yields and difficult purification protocols. Conventional routes frequently rely on harsh reaction conditions, such as extreme temperatures or the use of expensive transition metal catalysts that require rigorous removal to meet pharmaceutical purity standards. Furthermore, older methodologies might utilize unstable intermediates that degrade rapidly, leading to inconsistent batch-to-batch quality and significant material loss. The reliance on chromatographic purification for every step in traditional syntheses drastically increases production costs and limits the ability to scale up to commercial quantities. These inefficiencies create bottlenecks in the supply chain, causing delays in clinical trial material production and inflating the cost of goods sold for the final active pharmaceutical ingredient.
The Novel Approach
The methodology described in CN102775389A overcomes these historical challenges by employing a convergent strategy that maximizes atom economy and simplifies downstream processing. Instead of fragile intermediates, this route utilizes stable precursors that undergo efficient nucleophilic aromatic substitution and amide coupling. The process avoids the need for exotic catalysts, relying instead on standard reagents like thionyl chloride and triethylamine, which are cost-effective and easily sourced globally. A key innovation is the use of recrystallization rather than column chromatography for the final purification, a change that is pivotal for industrial scale-up. This approach not only enhances the purity profile of the final benzamide derivative but also significantly reduces solvent consumption and waste generation. By optimizing reaction times to between 4 to 8 hours per step, the process ensures a rapid turnover rate suitable for high-volume manufacturing environments.
Mechanistic Insights into Nucleophilic Substitution and Amide Coupling
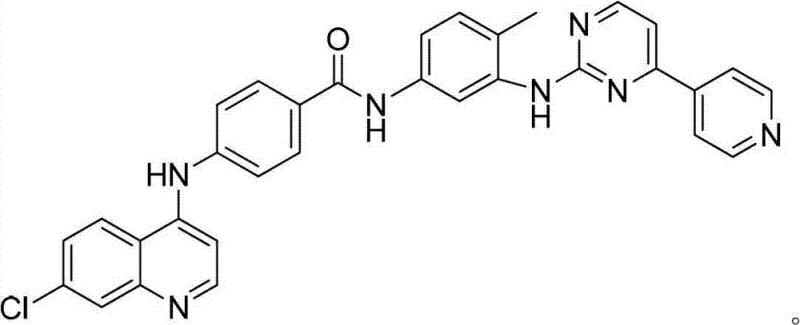
The core of this synthesis relies on a sequential activation strategy, beginning with the nucleophilic attack of an amine on a chloro-substituted heterocycle. In the first stage, p-aminobenzoic acid acts as a nucleophile, displacing the chlorine atom at the 4-position of the 4,7-dichloroquinoline ring system. This reaction is driven by reflux conditions in polar protic or aprotic solvents like isopropanol or tetrahydrofuran, which facilitate the solubility of both reactants and stabilize the transition state. The resulting intermediate, 4-(7-chloroquinoline-4-amino)benzoic acid, retains a carboxylic acid functionality that is crucial for the subsequent activation step. This specific regioselectivity is vital, as it ensures the 7-chloro group remains intact for potential further functionalization or maintains the specific pharmacophore required for kinase binding affinity.
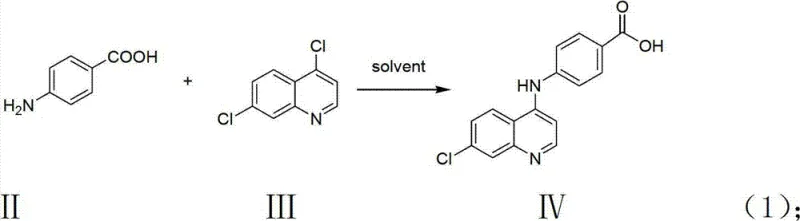
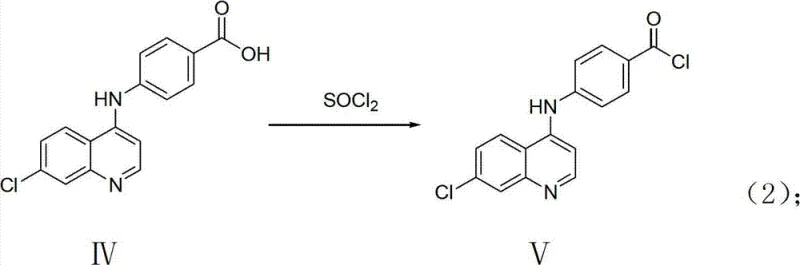
Following the formation of the quinoline-acid scaffold, the mechanism shifts to acyl chloride generation using thionyl chloride. This transformation converts the relatively unreactive carboxylic acid into a highly electrophilic acyl chloride, primed for nucleophilic attack by the amine component in the final step. The use of thionyl chloride is advantageous because the by-products, sulfur dioxide and hydrogen chloride, are gaseous and can be easily removed under reduced pressure, driving the equilibrium towards completion without leaving residual salts. In the final coupling stage, the acyl chloride reacts with the complex pyrimidine-amine derivative. The addition of triethylamine serves to scavenge the hydrochloric acid generated during amide bond formation, preventing the protonation of the amine nucleophile which would otherwise inhibit the reaction. This careful control of acidity ensures high conversion rates and minimizes the formation of hydrolysis by-products.
How to Synthesize N-Substituted Benzamide Derivatives Efficiently
Executing this synthesis requires precise control over stoichiometry and thermal parameters to maximize yield and purity. The process is divided into three distinct operational units: the initial condensation, the activation of the acid, and the final amide coupling. Each step has been optimized to balance reaction kinetics with safety considerations, particularly regarding the handling of thionyl chloride and the exothermic nature of the coupling reaction. Operators must adhere to strict temperature ranges, typically maintaining reflux for the initial steps and controlling the final coupling between 25°C and 50°C to prevent degradation of the sensitive pyrimidine moiety. The workup procedures involve strategic washing and filtration steps designed to remove inorganic salts and unreacted starting materials efficiently.
- Reflux p-aminobenzoic acid with 4,7-dichloroquinoline in organic solvent to form the quinoline-acid intermediate.
- React the intermediate acid with thionyl chloride under reflux to generate the reactive acyl chloride species.
- Couple the acyl chloride with the pyrimidine-amine derivative using triethylamine base, followed by recrystallization.
Commercial Advantages for Procurement and Supply Chain Teams
From a procurement perspective, this synthetic route offers substantial advantages due to its reliance on commodity chemicals and scalable unit operations. The starting materials, such as p-aminobenzoic acid and 4,7-dichloroquinoline, are commercially available in bulk quantities from multiple global suppliers, reducing the risk of single-source dependency. This diversity in the supply base ensures continuity of supply even during market fluctuations or geopolitical disruptions. Furthermore, the elimination of expensive transition metal catalysts removes the need for specialized scavenging resins and extensive heavy metal testing, which are significant cost drivers in API manufacturing. The simplified purification strategy, which favors crystallization over chromatography, translates directly into lower solvent costs and reduced waste disposal fees, contributing to a more sustainable and cost-effective production model.
- Cost Reduction in Manufacturing: The process achieves cost efficiency by utilizing thionyl chloride for activation, a reagent that is inexpensive and generates volatile by-products that do not require aqueous workup for removal. This eliminates the need for large volumes of water and the associated wastewater treatment costs. Additionally, the high yields reported in the patent examples, reaching up to 95% for the intermediate steps, minimize material loss and reduce the overall cost per kilogram of the final product. The ability to recycle solvents like tetrahydrofuran and isopropanol further enhances the economic viability of the process for large-scale campaigns.
- Enhanced Supply Chain Reliability: By avoiding proprietary or hard-to-source reagents, the manufacturing timeline is significantly de-risked. The reaction conditions are robust and tolerant to minor variations in raw material quality, which ensures consistent output. The use of standard glass-lined or stainless steel reactors for reflux and distillation steps means that the process can be transferred to almost any contract manufacturing organization without requiring specialized equipment investments. This flexibility allows for rapid scaling from pilot plant to commercial production, ensuring that lead times for clinical and commercial batches can be met reliably.
- Scalability and Environmental Compliance: The synthetic pathway is inherently scalable, as demonstrated by the straightforward workup procedures involving filtration and distillation. The reduction in solvent usage and the avoidance of heavy metals align with green chemistry principles, facilitating easier regulatory approval and environmental compliance. The process generates minimal hazardous waste, primarily consisting of organic solvent mixtures that can be distilled and recovered. This environmental profile is increasingly important for pharmaceutical companies aiming to reduce their carbon footprint and meet stringent sustainability goals imposed by global regulatory bodies.
Frequently Asked Questions (FAQ)
The following questions address common technical inquiries regarding the implementation and optimization of this synthesis route. Understanding these details is essential for process chemists and project managers evaluating the feasibility of adopting this technology for their pipeline. The answers are derived directly from the experimental data and specifications provided in the patent documentation, ensuring accuracy and relevance to real-world manufacturing scenarios.
Q: What are the critical reaction conditions for the acyl chloride formation step?
A: The process requires refluxing the benzoic acid intermediate with thionyl chloride for 2-4 hours, followed by reduced pressure distillation to remove excess reagent, ensuring high conversion to the acyl chloride.
Q: How is product purity maintained during the final coupling step?
A: Purity is ensured by controlling the reaction temperature between 25°C-50°C, removing salt by-products via suction filtration, and performing a final recrystallization using dichloromethane and methanol.
Q: What solvents are compatible with this synthesis route?
A: The protocol supports versatile solvent systems including isopropanol, ethanol, tetrahydrofuran, or pyridine for the initial substitution, offering flexibility for large-scale optimization.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable N-Substituted Benzamide Derivative Supplier
At NINGBO INNO PHARMCHEM, we possess the technical expertise and infrastructure to bring complex synthetic routes like the one described in CN102775389A from the laboratory bench to full commercial production. Our team has extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your supply needs are met with precision and consistency. We operate stringent purity specifications and maintain rigorous QC labs equipped with advanced analytical instrumentation to verify the identity and quality of every batch. Our commitment to quality assurance means that we can deliver high-purity pharmaceutical intermediates that meet the demanding requirements of global regulatory agencies.
We invite you to collaborate with us to optimize this synthesis for your specific application needs. Our technical procurement team is ready to provide a Customized Cost-Saving Analysis tailored to your volume requirements and timeline. Please contact us to request specific COA data and route feasibility assessments, and let us demonstrate how our manufacturing capabilities can accelerate your drug development program while reducing overall production costs.