Advanced Manufacturing of 2'-Substituted Pyrimidine Nucleosides for Next-Gen Nucleic Acid Therapeutics
Advanced Manufacturing of 2'-Substituted Pyrimidine Nucleosides for Next-Gen Nucleic Acid Therapeutics
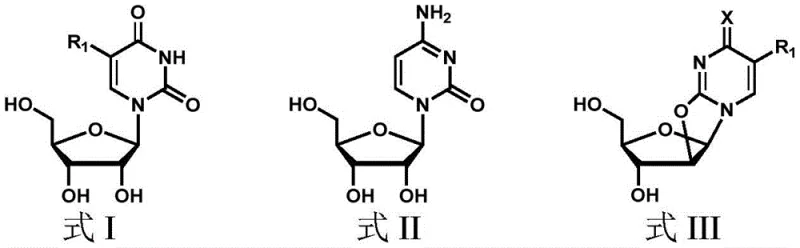
The rapidly evolving landscape of nucleic acid therapeutics demands increasingly sophisticated chemical building blocks that offer superior stability and bioavailability. Patent CN114369124A, published in April 2022, introduces a groundbreaking preparation method for 2'-substituted pyrimidine nucleosides, addressing critical bottlenecks in the synthesis of these vital pharmaceutical intermediates. This technology represents a significant leap forward in the chemical synthesis of modified nucleosides, which serve as the fundamental monomers for antisense oligonucleotides (ASOs), siRNA, and mRNA therapies. By optimizing the ring-opening strategy of anhydro nucleosides, this invention provides a pathway to high-purity products with exceptional yield consistency, directly impacting the cost and reliability of the global supply chain for nucleic acid drugs.
The core innovation lies in a strategic four-step sequence that fundamentally alters the physical properties of the reaction intermediates to favor desired product formation. Unlike traditional methods that struggle with solubility and side reactions, this approach employs a protected anhydropyrimidine nucleoside as the key substrate for the ring-opening step. This subtle yet powerful modification ensures that the reaction proceeds under milder conditions while effectively suppressing the formation of problematic dimers. For R&D directors and process chemists, this patent offers a robust blueprint for synthesizing complex 2'-modified nucleosides such as 2'-O-methyluridine and 2'-O-methoxyethylcytidine with industrial-grade efficiency.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of 2'-hydroxyl substituted pyrimidine nucleosides has relied heavily on the direct ring-opening of dehydrated uridine or cytidine derivatives. While foundational methods developed by researchers like Saroj K. Roy demonstrated feasibility, they were plagued by inherent chemical limitations that hindered large-scale adoption. The primary challenge stems from the poor solubility of unprotected anhydro nucleosides in the organic solvents required for alkylation, often necessitating harsh reaction conditions or exotic reagents like methoxyethylaluminum. Furthermore, the lack of protection at the 5'-position leaves the molecule vulnerable to intermolecular reactions, leading to the formation of dimers and other oligomeric impurities that are notoriously difficult to separate. These side reactions not only depress the overall yield, often capping it below optimal levels, but also create a complex impurity profile that requires extensive and costly downstream purification processes to meet pharmaceutical standards.
The Novel Approach
The methodology disclosed in CN114369124A circumvents these historical hurdles through a clever manipulation of the substrate's steric and electronic environment prior to the critical ring-opening event. By introducing a bulky 5'-O-di-p-methoxytrityl (DMTr) protecting group before the ring-opening step, the process dramatically enhances the solubility of the anhydro intermediate in standard organic solvents like dichloromethane and alcohols. This solubility improvement allows for more homogeneous reaction conditions, facilitating better heat and mass transfer which is essential for scale-up. Moreover, the steric bulk of the DMTr group physically shields the 5'-position, effectively preventing the nucleophilic attack that leads to dimerization. The result is a cleaner reaction profile with significantly higher yields, as evidenced by the patent examples where yields consistently exceed 90% for the ring-opening step.
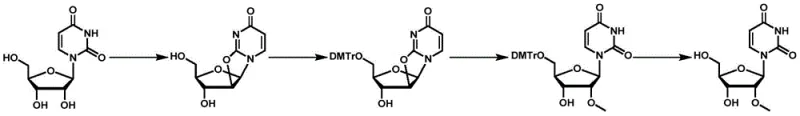
Mechanistic Insights into Magnesium Alkoxide-Mediated Ring Opening
The heart of this synthetic innovation is the third step: the regioselective ring-opening of the protected anhydro nucleoside using magnesium alkoxide. In this mechanism, the magnesium alkoxide acts as both a base and a nucleophile source. The magnesium cation coordinates with the oxygen atoms of the furanose ring and the carbonyl groups of the pyrimidine base, activating the epoxide-like 2,2'-anhydro bond towards nucleophilic attack. The alkoxide ion then attacks the C2' position, cleaving the anhydro bridge and installing the desired 2'-substituent (such as a methyl or methoxyethyl group). Because the 5'-hydroxyl is masked by the DMTr group, the alkoxide is directed exclusively to the 2'-position, ensuring high regioselectivity. This coordination chemistry is far more controlled than the aluminum-mediated processes of the past, reducing the risk of over-alkylation or degradation of the sensitive glycosidic bond.
From an impurity control perspective, this mechanism is exceptionally clean. The avoidance of dimer formation is the most significant advantage, as dimers share similar polarity and chromatographic behavior to the monomer, making them a nightmare for purification teams. By preventing their formation at the source, the process simplifies the workup procedure significantly. The patent details that the reaction can be quenched simply by neutralizing excess magnesium alkoxide with acetic acid, followed by standard aqueous washes. This eliminates the need for complex quenching protocols often required by organometallic reagents. Furthermore, the final deprotection step using mild acids like acetic acid or formic acid ensures that the acid-labile DMTr group is removed without damaging the newly formed 2'-ether linkage or the glycosidic bond, resulting in a final product with minimal inorganic salt residue, a critical quality attribute for API intermediates.
How to Synthesize 2'-O-Methyluridine Efficiently
The synthesis of 2'-O-methyluridine serves as the flagship example of this technology, demonstrating the practical application of the four-step sequence. The process begins with the dehydration of commercially available uridine using diphenyl carbonate, followed by selective protection of the 5'-hydroxyl group. The protected intermediate is then subjected to ring-opening with magnesium methoxide, and finally, the protecting group is removed to yield the target nucleoside. This route is notable for its operational simplicity and the use of readily available reagents, making it highly attractive for process development teams looking to establish a reliable supply chain. The detailed standardized synthesis steps for this specific transformation are outlined in the guide below.
- Dehydration of uridine or cytidine using diphenyl carbonate and a base to form the 2,2'-anhydro intermediate.
- Selective 5'-position protection using bis-p-methoxytriphenylchloromethane (DMTr-Cl) to improve substrate solubility.
- Ring-opening reaction using magnesium alkoxide in alcohol solvent to introduce the 2'-substituent without dimer formation.
- Acidic deprotection to remove the 5'-protecting group and isolate the final 2'-substituted pyrimidine nucleoside product.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the technical advantages of this patent translate directly into tangible business benefits regarding cost, reliability, and scalability. The shift from unprotected to protected intermediates fundamentally changes the economics of manufacturing 2'-substituted nucleosides. By eliminating the formation of hard-to-remove dimer impurities, the process reduces the burden on purification units, lowering solvent consumption and processing time. This efficiency gain is crucial for maintaining competitive pricing in the high-volume nucleic acid drug market. Furthermore, the use of magnesium alkoxides, which are cheaper and easier to handle than aluminum-based reagents, contributes to a reduction in raw material costs and safety hazards associated with pyrophoric materials.
- Cost Reduction in Manufacturing: The streamlined purification process resulting from the high selectivity of this method leads to substantial cost savings. By avoiding the generation of dimer impurities, manufacturers can reduce the number of chromatography columns or recrystallization cycles required to reach pharmaceutical purity. This not only saves on consumables like silica gel and solvents but also increases the throughput of existing manufacturing facilities. Additionally, the high yields reported in the patent examples mean that less starting material is wasted, maximizing the return on investment for expensive nucleoside precursors and driving down the cost per kilogram of the final active ingredient.
- Enhanced Supply Chain Reliability: The robustness of this synthetic route ensures a more stable supply of critical nucleoside monomers. The reaction conditions are mild and tolerant, reducing the risk of batch failures due to minor fluctuations in temperature or reagent quality. This reliability is paramount for pharmaceutical companies that require consistent quality for regulatory filings. The ability to produce intermediates with consistently low residue on ignition (below 0.1%) means that downstream customers face fewer quality disputes and delays, fostering a more resilient and trustworthy supply partnership.
- Scalability and Environmental Compliance: This method is inherently designed for commercial scale-up, utilizing solvents and reagents that are common in the fine chemical industry. The avoidance of heavy metal catalysts simplifies waste treatment and disposal, aligning with increasingly stringent environmental regulations. The process generates less hazardous waste compared to traditional organometallic routes, reducing the environmental footprint of production. This compliance advantage minimizes the risk of regulatory shutdowns and ensures long-term sustainability for the manufacturing site, securing the supply chain against future environmental policy changes.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. These answers are derived directly from the experimental data and claims presented in the patent documentation, providing a clear understanding of the method's capabilities and limitations. Understanding these details is essential for technical teams evaluating the feasibility of adopting this route for their specific product portfolios.
Q: Why is the 5'-protection step critical in this synthesis route?
A: The 5'-protection step significantly improves the solubility of the anhydro nucleoside substrate in organic solvents. More importantly, it prevents the formation of dimers during the subsequent ring-opening reaction, which is a common side reaction in conventional unprotected routes, thereby drastically improving overall yield and purity.
Q: What are the typical purity levels achieved with this method?
A: According to the patent examples, the final products consistently achieve HPLC purity greater than 98%. Additionally, the process effectively removes inorganic salts, resulting in extremely low residue on ignition (e.g., 0.05%), which is critical for pharmaceutical grade intermediates.
Q: Is this process suitable for large-scale industrial production?
A: Yes, the method is designed for industrial scalability. It utilizes mild reaction conditions (temperatures ranging from 60°C to 150°C) and avoids the use of difficult-to-remove transition metal catalysts. The high yields (often exceeding 90% per step) and robust impurity profile make it highly suitable for commercial scale-up.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 2'-Substituted Pyrimidine Nucleoside Supplier
At NINGBO INNO PHARMCHEM, we recognize the pivotal role that high-quality nucleoside monomers play in the development of life-saving nucleic acid therapies. Our team of expert process chemists has thoroughly analyzed the methodology described in CN114369124A and possesses the extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production. We are committed to delivering these complex intermediates with stringent purity specifications, utilizing our rigorous QC labs to ensure every batch meets the exacting standards required for GMP manufacturing. Our facility is equipped to handle the specific solvent systems and protection/deprotection chemistries required by this advanced route, ensuring a seamless transition from lab scale to industrial volume.
We invite pharmaceutical partners to collaborate with us to leverage this innovative synthesis technology for their pipeline projects. By partnering with us, you gain access to a Customized Cost-Saving Analysis that quantifies the potential efficiencies of switching to this protected intermediate route. We encourage you to contact our technical procurement team today to request specific COA data from our pilot batches and to discuss detailed route feasibility assessments tailored to your specific 2'-substituted nucleoside requirements. Let us help you secure a stable, high-quality supply of these critical building blocks for your next-generation therapeutics.