Scalable Metal-Free Synthesis of Monofluorinated 4H-Pyran Derivatives for Advanced Drug Discovery
The pharmaceutical and fine chemical industries are constantly seeking robust, scalable, and cost-effective methodologies for constructing fluorinated heterocyclic scaffolds, which are pivotal in modern drug design. Patent CN115043807A introduces a groundbreaking approach for the synthesis of monofluorinated 4H-pyran compounds, addressing critical bottlenecks in traditional heterocycle construction. This technology leverages a base-catalyzed annulation strategy between beta-trifluoromethyl-1,3-enyne compounds and various 1,3-dicarbonyl substrates, such as acetylacetone, ethyl acetoacetate, or dimedone. Unlike conventional routes that often rely on harsh conditions or precious metal catalysis, this invention operates under remarkably mild parameters, utilizing inexpensive inorganic bases like potassium phosphate in polar aprotic solvents. The strategic incorporation of a single fluorine atom into the 4H-pyran core significantly enhances the lipophilicity and metabolic stability of the resulting molecules, making them highly attractive candidates for medicinal chemistry programs targeting neurodegenerative diseases and calcium channel modulation. For R&D directors and procurement specialists alike, this patent represents a significant leap forward in accessing high-value fluorinated building blocks with superior purity profiles and reduced environmental footprints.
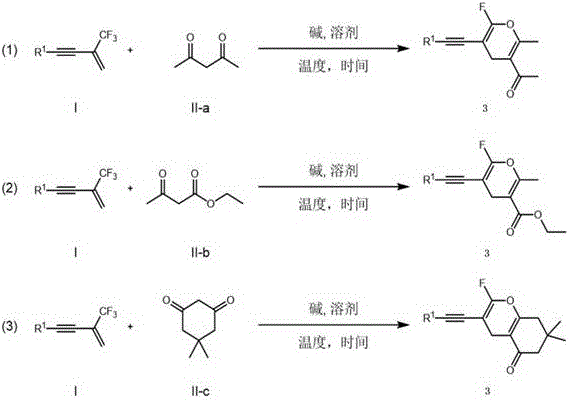
The limitations of conventional methods for synthesizing fluorinated pyran derivatives often stem from their reliance on complex multi-step sequences or the use of toxic transition metal catalysts that necessitate rigorous purification protocols to meet regulatory standards for residual metals in active pharmaceutical ingredients (APIs). Traditional approaches may involve high-temperature cyclizations that compromise sensitive functional groups or require stoichiometric amounts of expensive reagents, driving up the cost of goods sold (COGS) and complicating supply chain logistics. Furthermore, many existing methods struggle with substrate generality, failing to accommodate diverse electronic environments on the aromatic rings, which limits their utility in the rapid exploration of structure-activity relationships (SAR). In contrast, the novel approach detailed in this patent circumvents these issues by employing a direct, one-pot cyclization mechanism driven by simple base catalysis. This eliminates the need for transition metals entirely, thereby removing the costly and time-consuming heavy metal scavenging steps typically required in GMP manufacturing. The reaction conditions are exceptionally mild, typically proceeding at 50°C, which not only reduces energy consumption but also preserves the integrity of labile functional groups such as esters, nitriles, and halides that are frequently present in advanced drug intermediates.
Mechanistic Insights into Base-Catalyzed Defluorinative Cyclization
The mechanistic pathway of this transformation is a sophisticated cascade of nucleophilic attacks and elimination events that elegantly constructs the fluorinated heterocyclic core with high atom economy. The process initiates with the deprotonation of the active methylene group in the 1,3-dicarbonyl compound by the base, generating a resonance-stabilized carbanion species. This nucleophile then undergoes a conjugate addition to the terminal alkene of the beta-trifluoromethyl-1,3-enyne substrate, forming a new carbon-carbon bond and a stabilized anionic intermediate. Crucially, the presence of the trifluoromethyl group facilitates a subsequent beta-fluorine elimination, driven by the electron-withdrawing nature of the adjacent groups, which generates a gem-difluoro alkene intermediate. This species rapidly tautomerizes to an enol form, positioning the oxygen nucleophile for an intramolecular attack on the electrophilic carbon center. The final step involves a defluorinative ring closure that aromatizes the pyran system, expelling a fluoride ion and yielding the stable monofluorinated 4H-pyran product. This intricate sequence ensures high regioselectivity and minimizes the formation of side products, providing R&D teams with a predictable and reliable route to complex scaffolds.
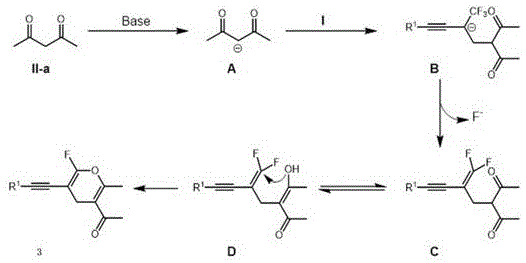
Understanding the impurity control mechanisms inherent in this chemistry is vital for ensuring the high purity required for pharmaceutical applications. The mild basic conditions and the specific electronic activation provided by the trifluoromethyl group suppress common side reactions such as polymerization of the enyne substrate or over-alkylation of the dicarbonyl partner. The reaction kinetics favor the desired cyclization pathway over competing intermolecular condensations, largely due to the entropic advantage of the intramolecular ring-closing step. Furthermore, the use of polar aprotic solvents like DMF enhances the solubility of the ionic intermediates, preventing precipitation that could lead to incomplete reactions or difficult-to-remove occluded impurities. For quality control laboratories, this translates to cleaner crude reaction mixtures that simplify downstream purification, often allowing for high recovery yields via standard column chromatography or even crystallization. The robustness of the mechanism against varying electronic substituents on the aromatic ring—ranging from electron-donating methoxy groups to electron-withdrawing cyano and halide groups—ensures consistent impurity profiles across a wide library of analogs, facilitating faster scale-up and regulatory filing.
How to Synthesize Monofluorinated 4H-Pyran Efficiently
To implement this synthesis effectively in a laboratory or pilot plant setting, operators should adhere to the optimized conditions established in the patent examples, which balance reaction rate with product quality. The standard protocol involves charging a reaction vessel with the beta-trifluoromethyl-1,3-enyne starting material and the chosen 1,3-dicarbonyl compound in a molar ratio of approximately 1:1.5 to drive the reaction to completion while minimizing excess reagent waste. Potassium phosphate is added as the base at 150 mol% relative to the enyne, ensuring sufficient catalytic turnover without inducing excessive decomposition. The mixture is suspended in anhydrous DMF and heated to 50°C with vigorous stirring for 4 hours. Upon completion, the reaction is quenched with water, extracted with an organic solvent such as ethyl acetate, and the combined organic layers are concentrated. The crude product is typically purified by silica gel column chromatography to afford the target monofluorinated 4H-pyran as a high-purity solid or oil. Detailed standardized synthesis steps are provided in the guide below.
- Combine beta-trifluoromethyl-1,3-enyne compound I with a 1,3-dicarbonyl substrate (such as acetylacetone or ethyl acetoacetate) in a reaction vessel.
- Add potassium phosphate (K3PO4) as the base catalyst and N,N-dimethylformamide (DMF) as the solvent to the mixture.
- Stir the reaction mixture at 50°C for approximately 4 hours, then quench, extract, and purify via column chromatography to isolate the target monofluorinated 4H-pyran.
Commercial Advantages for Procurement and Supply Chain Teams
From a commercial perspective, this technology offers substantial advantages for procurement managers and supply chain heads looking to optimize the sourcing of fluorinated intermediates. The elimination of transition metal catalysts is a primary driver for cost reduction in fine chemical manufacturing, as it removes the necessity for purchasing expensive palladium, copper, or rhodium complexes and the associated ligands. Moreover, it obviates the need for specialized metal scavenger resins and the extensive analytical testing required to certify low residual metal levels, significantly shortening the production cycle time. The use of commodity chemicals like potassium phosphate and DMF ensures a stable and resilient supply chain, reducing the risk of disruptions caused by the scarcity of specialized reagents. The mild reaction temperature of 50°C also contributes to lower energy costs compared to high-temperature reflux processes, aligning with sustainability goals and reducing the overall carbon footprint of the manufacturing process. These factors collectively enhance the economic viability of producing these high-value intermediates at scale.
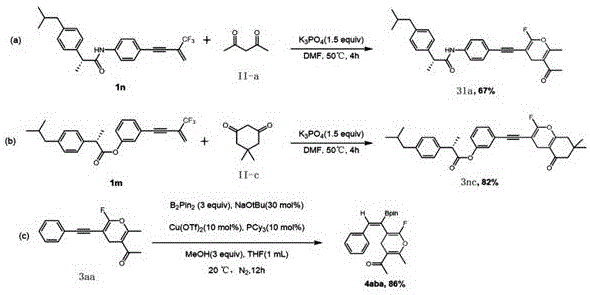
Enhanced supply chain reliability is another critical benefit, as the simplicity of the reaction setup allows for easy technology transfer between different manufacturing sites without the need for specialized corrosion-resistant equipment often required for harsh acidic or basic conditions. The broad substrate scope demonstrated in the patent, which includes successful derivatization of complex drug-like molecules such as ibuprofen analogs, indicates that this method can be seamlessly integrated into existing pipelines for API intermediate production. This versatility reduces the lead time for high-purity pharmaceutical intermediates by allowing chemists to access diverse analogs from a common set of starting materials without re-optimizing reaction conditions for each new substrate. Additionally, the high yields reported across various examples suggest minimal material loss, maximizing the throughput of raw materials and reducing waste disposal costs. The ability to perform late-stage functionalization, such as the copper-catalyzed borylation shown in the patent data, further extends the utility of these intermediates, enabling rapid diversification for SAR studies without restarting the synthesis from scratch.
- Cost Reduction in Manufacturing: The complete absence of precious metal catalysts drastically lowers raw material costs and eliminates expensive purification steps associated with metal removal. By utilizing inexpensive inorganic bases and common solvents, the overall cost of goods is significantly optimized, making the production of fluorinated heterocycles more economically accessible for large-scale campaigns.
- Enhanced Supply Chain Reliability: Reliance on widely available commodity chemicals rather than specialized organometallic reagents ensures a stable supply chain with minimal risk of vendor lock-in or shortage. The robust nature of the reaction conditions allows for flexible manufacturing scheduling and easier scaling from gram to ton quantities without compromising quality or consistency.
- Scalability and Environmental Compliance: The mild operating conditions and high atom economy of this process align with green chemistry principles, reducing hazardous waste generation and energy consumption. This facilitates smoother regulatory approvals and environmental compliance audits, while the simple workup procedures enable efficient scale-up to commercial production volumes with minimal engineering constraints.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology, based on the detailed experimental data provided in the patent documentation. These insights are designed to clarify the operational feasibility and strategic value of adopting this metal-free route for your specific project needs. Understanding these nuances will help stakeholders make informed decisions about integrating this methodology into their current development workflows.
Q: Does this synthesis method require expensive transition metal catalysts?
A: No, the method described in patent CN115043807A utilizes inexpensive inorganic bases like potassium phosphate, completely eliminating the need for costly transition metal catalysts and subsequent heavy metal removal steps.
Q: What is the typical reaction temperature and time for this cyclization?
A: The reaction proceeds under mild conditions, typically requiring a temperature of 50°C and a reaction time of approximately 4 hours in DMF solvent, ensuring energy efficiency and operational safety.
Q: Can this method be applied to complex drug-like molecules?
A: Yes, the protocol demonstrates excellent functional group tolerance, successfully synthesizing derivatives containing ibuprofen scaffolds and allowing for further late-stage functionalization such as borylation.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Monofluorinated 4H-Pyran Supplier
At NINGBO INNO PHARMCHEM, we recognize the transformative potential of this metal-free synthesis technology for accelerating drug discovery programs. As a leading CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your transition from benchtop to market is seamless and efficient. Our state-of-the-art facilities are equipped with rigorous QC labs capable of meeting stringent purity specifications, guaranteeing that every batch of monofluorinated 4H-pyan intermediates meets the highest international standards. We understand the critical importance of supply continuity in the pharmaceutical sector and are committed to providing a reliable source of these high-value building blocks to support your long-term development goals.
We invite you to engage with our technical procurement team to discuss how this innovative synthesis route can be tailored to your specific project requirements. By partnering with us, you gain access to a Customized Cost-Saving Analysis that evaluates the economic benefits of switching to this metal-free protocol for your specific molecule. We encourage you to request specific COA data and route feasibility assessments to validate the performance of these intermediates in your downstream processes. Let us collaborate to unlock the full potential of fluorinated heterocycles in your next breakthrough therapy.