Advanced Manufacturing of Dolastatin 10 Intermediates via Stereoselective Beta-Addition
The pharmaceutical industry continuously seeks robust and scalable synthetic routes for complex peptide mimetics, particularly those serving as critical building blocks for anticancer agents. Patent CN101076515A discloses a novel and improved process for the manufacture of 3-pyrrolidin-2-yl-propionic acid derivatives, specifically compounds of formula (I), which serve as valuable intermediates in the synthesis of Dolastatin 10 analogues. Dolastatin 10 is a potent antimitotic peptide originally isolated from the marine mollusc Dolabella auricularia, known for inhibiting tubulin polymerization. While previous methods, such as those disclosed in WO 03/008378, provided access to these structures, they suffered from low yields and cumbersome purification steps involving chromatography to separate diastereomeric mixtures formed during the beta-addition reaction. The present invention addresses these critical bottlenecks by introducing two distinct reaction procedures, A and B, which leverage specific proton sources and crystallization techniques to achieve superior diastereomer ratios and overall yields without the need for extensive chromatographic purification.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditional synthetic pathways for generating the dolaproine moiety found in Dolastatin 10 derivatives often rely on the conjugate addition of thiols to alpha,beta-unsaturated esters derived from proline. A significant challenge in this transformation is the control of stereochemistry at the newly formed chiral centers. In prior art methods, the beta-addition step typically results in a complex mixture of diastereoisomers. Separating these isomers to obtain the pharmacologically active configuration usually requires preparative high-performance liquid chromatography (HPLC) or flash column chromatography. These separation techniques are not only time-consuming and labor-intensive but also result in substantial material loss, drastically reducing the overall process yield. Furthermore, the use of large volumes of solvents and silica gel for chromatography increases the environmental footprint and manufacturing costs, making such processes less attractive for commercial scale-up where efficiency and waste reduction are paramount concerns for supply chain sustainability.
The Novel Approach
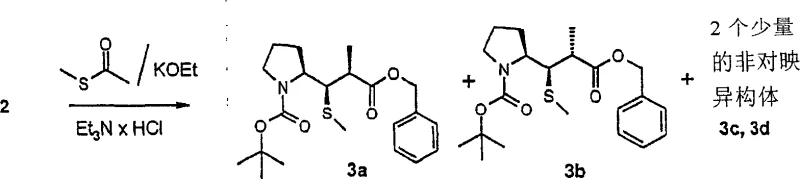
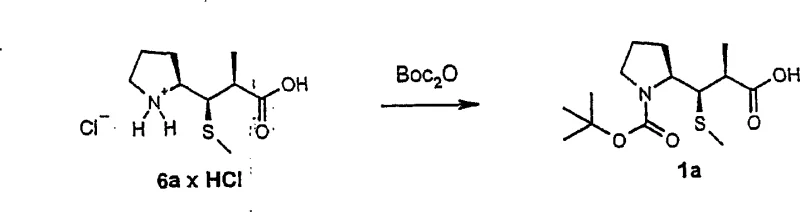
The methodology described in CN101076515A represents a significant technological leap by optimizing the reaction conditions to favor the formation of the desired diastereomer directly. The core innovation lies in the modification of the beta-addition step. By employing triethylammonium chloride (Et3N x HCl) as a proton source in the presence of a potassium base, the process achieves a marked improvement in diastereoselectivity. Experimental data within the patent indicates that this specific additive can shift the diastereomer ratio (dr) to approximately 90:10 or 89:11, a substantial improvement over the 70:30 ratios observed with other proton sources. Additionally, the patent introduces clever purification strategies that bypass chromatography entirely. Procedure A utilizes hydrogenolysis followed by the formation of specific ammonium salts (e.g., with dicyclohexylamine) which crystallize with high diastereomeric purity. Procedure B employs acid-induced precipitation of a hydrochloride salt followed by re-protection. Both pathways transform a difficult separation problem into a manageable crystallization process, significantly enhancing the economic viability of the synthesis.
Mechanistic Insights into Stereoselective Beta-Addition
The heart of this synthetic advancement is the stereoselective conjugate addition of the thiol group to the unsaturated proline derivative. The reaction begins with a Wittig olefination of Boc-L-prolinal to generate the alpha,beta-unsaturated ester, typically yielding the E-isomer with high selectivity (E/Z > 95:5). The subsequent addition of the thiol nucleophile is where the stereochemical fate is determined. In standard Michael additions, the approach of the nucleophile can occur from either face of the planar double bond, leading to a mixture of diastereomers. The patent reveals that the nature of the proton source used to quench the enolate intermediate plays a crucial role in this selectivity. Triethylammonium chloride appears to provide a specific steric or electronic environment that favors the formation of the (1R, 2S) configuration over the (1R, 2R) isomer. This suggests a structured transition state where the ammonium ion may coordinate with the carbonyl oxygen or the sulfur atom, directing the protonation step to lock in the desired stereochemistry. This mechanistic understanding allows chemists to fine-tune the reaction conditions to maximize the yield of the target isomer before any purification even begins.
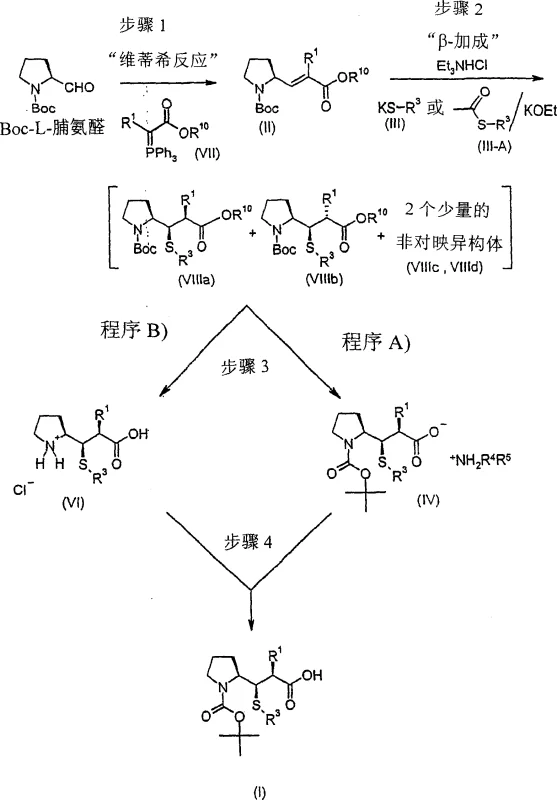
Following the beta-addition, the crude mixture contains the desired product along with minor diastereomers. The patent details how these impurities are managed not by separation, but by enrichment. In Procedure A, the benzyl ester is cleaved via hydrogenolysis to reveal the free carboxylic acid. This acid is then treated with a bulky amine, such as dicyclohexylamine. The resulting ammonium salt of the major diastereomer has significantly lower solubility in solvents like tert-butyl methyl ether compared to the salts of the minor diastereomers. Consequently, upon cooling, the desired salt crystallizes out in high purity (dr > 97:3), leaving the impurities in the mother liquor. This phenomenon, known as crystallization-induced diastereomer enrichment, is a powerful tool in process chemistry. Similarly, Procedure B exploits the solubility differences of hydrochloride salts in ethyl acetate. By treating the crude tert-butyl ester mixture with anhydrous HCl, the desired diastereomer precipitates as a hydrochloride salt, which can be filtered and washed to remove impurities before being re-protected with Boc anhydride. These mechanisms ensure that the final product meets stringent purity specifications required for pharmaceutical intermediates.
How to Synthesize 3-pyrrolidin-2-yl-propionic acid derivatives Efficiently
The synthesis of these high-value intermediates can be executed via two primary workflows depending on the protecting group strategy employed. Both routes start with the preparation of the unsaturated precursor via Wittig reaction. The critical divergence occurs in the workup and purification phases. For manufacturers prioritizing the removal of benzyl groups early, Procedure A offers a robust path involving catalytic hydrogenation and salt crystallization. For those preferring to maintain acid-labile protecting groups throughout, Procedure B provides an alternative utilizing acid precipitation. Both methods eliminate the need for silica gel chromatography, relying instead on filtration and crystallization unit operations which are far more amenable to large-scale reactor setups. The detailed standardized synthesis steps for optimizing these parameters, including specific solvent swaps and temperature profiles for crystallization, are outlined in the guide below.
- Perform Wittig reaction on Boc-L-prolinal with appropriate ylide to form alpha,beta-unsaturated ester.
- Execute beta-addition of alkyl-mercaptan using potassium ethoxide and triethylammonium chloride to enhance diastereoselectivity.
- Purify the resulting mixture via hydrogenolysis and amine salt formation (Procedure A) or acid precipitation and re-protection (Procedure B).
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the transition from chromatography-dependent synthesis to crystallization-based purification represents a transformative shift in cost structure and operational reliability. The elimination of chromatographic columns removes a major bottleneck in production throughput. Chromatography is inherently a batch process with limited capacity, requiring significant downtime for column packing, loading, elution, and regeneration. By replacing this with crystallization, the manufacturing process becomes continuous-friendly and significantly faster. This reduction in processing time directly translates to increased plant capacity and the ability to fulfill large orders with shorter lead times. Furthermore, the reduction in solvent consumption and solid waste (silica gel) aligns with modern green chemistry initiatives, potentially lowering waste disposal costs and simplifying regulatory compliance regarding environmental emissions.
- Cost Reduction in Manufacturing: The primary driver for cost optimization in this process is the removal of chromatographic purification. Chromatography resins and the associated solvents are expensive consumables that contribute heavily to the Cost of Goods Sold (COGS). By achieving high diastereomeric purity through selective crystallization, the process drastically reduces material costs. Additionally, the improved yield resulting from minimized material loss during purification means that less starting material is required to produce the same amount of final product. The use of commercially available reagents like triethylammonium chloride and standard bases like potassium ethoxide ensures that raw material costs remain stable and predictable, avoiding the volatility associated with specialized chiral catalysts or exotic reagents.
- Enhanced Supply Chain Reliability: Supply chain resilience is bolstered by the robustness of the chemical transformations described. The reactions utilize standard conditions (e.g., reflux in THF, room temperature crystallizations) that do not require extreme cryogenic temperatures or high-pressure equipment beyond standard hydrogenation reactors. This compatibility with existing multi-purpose chemical manufacturing infrastructure means that production can be easily scaled or transferred between different facilities without significant capital investment. The ability to produce intermediates with consistent quality (high dr and ee) reduces the risk of batch failures and rejections, ensuring a steady flow of materials for downstream API synthesis. Reliable availability of key intermediates is critical for maintaining the production schedules of life-saving oncology drugs.
- Scalability and Environmental Compliance: Scaling a process that relies on crystallization is fundamentally easier than scaling one dependent on chromatography. Crystallization kinetics are well-understood and can be modeled effectively from laboratory to pilot to commercial scale. The process described generates less hazardous waste, as it avoids the disposal of spent silica gel contaminated with organic compounds. The solvents used, such as ethyl acetate, heptane, and tert-butyl methyl ether, are commonly recovered and recycled in modern pharmaceutical plants, further minimizing the environmental footprint. This alignment with sustainable manufacturing practices not only reduces costs but also enhances the corporate social responsibility profile of the supply chain, a factor increasingly important to global pharmaceutical partners.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this novel synthesis route. Understanding these details is essential for R&D teams evaluating the feasibility of technology transfer and for procurement specialists assessing the long-term viability of the supply source. The answers are derived directly from the experimental data and claims presented in the patent documentation, ensuring accuracy and relevance to real-world manufacturing scenarios.
Q: How does the novel process improve diastereomer ratios compared to prior art?
A: The process utilizes triethylammonium chloride as a specific proton source during the beta-addition step, which significantly enhances diastereoselectivity (up to 90:10 dr) compared to conventional proton sources, reducing the need for chromatographic separation.
Q: What are the key purification strategies described in the patent?
A: The patent outlines two main strategies: Procedure A uses hydrogenolysis followed by crystallization of an ammonium salt (e.g., with dicyclohexylamine) to enrich purity, while Procedure B utilizes direct precipitation of a hydrochloride salt followed by N-Boc re-protection.
Q: Is this process suitable for large-scale commercial production?
A: Yes, the avoidance of laborious chromatography and the reliance on crystallization-induced diastereomer enrichment make this method highly scalable and economically viable for industrial manufacturing of Dolastatin 10 analogues.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 3-pyrrolidin-2-yl-propionic acid derivative Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of high-quality intermediates in the development of next-generation anticancer therapies. Our technical team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the sophisticated stereoselective techniques described in CN101076515A can be successfully translated from the laboratory bench to industrial reactors. We maintain stringent purity specifications and operate rigorous QC labs equipped with advanced chiral HPLC and NMR capabilities to verify the diastereomeric and enantiomeric excess of every batch. Our commitment to quality assurance guarantees that the Dolastatin 10 intermediates we supply meet the exacting standards required for clinical and commercial drug substance manufacturing.
We invite potential partners to engage with our technical procurement team to discuss how this optimized process can benefit your specific supply chain needs. By leveraging our manufacturing expertise, you can achieve significant efficiencies in your API production. Please contact us to request a Customized Cost-Saving Analysis tailored to your volume requirements. We are prepared to provide specific COA data and comprehensive route feasibility assessments to demonstrate how our advanced synthesis capabilities can support your long-term strategic goals in the oncology sector.