Advanced Sitagliptin Free Base Manufacturing: Overcoming Legacy Process Limitations for Commercial Scale
Advanced Sitagliptin Free Base Manufacturing: Overcoming Legacy Process Limitations for Commercial Scale
The pharmaceutical industry continuously seeks robust synthetic routes that balance high purity with operational safety and cost efficiency. A pivotal advancement in this domain is detailed in Chinese Patent CN113149991A, which discloses a novel synthesis method for sitagliptin free base and its phosphate monohydrate salt. This technology addresses critical bottlenecks found in earlier generations of manufacturing processes, specifically targeting the elimination of hazardous reagents and the simplification of purification steps. By transitioning from solvent-intensive protocols to a streamlined in-situ process, this method offers a compelling value proposition for reliable sitagliptin intermediate suppliers aiming to optimize their production lines. The core innovation lies in the strategic replacement of dimethylformamide (DMF) and the utilization of HOBt hydrate, which collectively enhance reaction safety and facilitate easier solvent recovery.
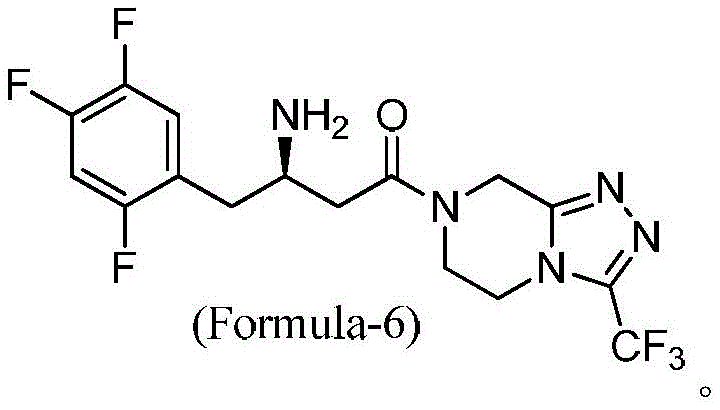
The significance of this patent extends beyond mere chemical transformation; it represents a shift towards greener and more economically viable API manufacturing. Traditional routes often suffered from low yields due to the necessity of chromatographic purification or the generation of stubborn impurities during the deprotection phase. The methodology described in CN113149991A circumvents these issues by employing a one-pot strategy that converts the protected amino acid directly to the final free base with minimal isolation steps. For procurement managers and supply chain directors, this translates to a reduction in unit operations and a significant decrease in the consumption of auxiliary materials. The ability to achieve an HPLC purity of greater than 99% without resorting to preparative thin-layer chromatography marks a substantial leap forward in process intensification.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Prior art technologies, such as those disclosed in US Patent 6699871B2 and International Application WO2009064476A1, present several formidable challenges for large-scale production. The earlier US patent describes a condensation reaction that relies on dried HOBt and methylene chloride, yielding the intermediate carbamate in a mere 33% after purification by preparative thin-layer chromatography. This reliance on chromatography is a major bottleneck, as it is inherently batch-limited, labor-intensive, and difficult to scale for multi-ton production campaigns. Furthermore, the use of anhydrous HOBt introduces a severe safety hazard due to its potential explosiveness, necessitating specialized handling equipment and rigorous safety protocols that drive up operational expenditures.
Similarly, the process outlined in WO2009064476A1 attempts to improve yields but introduces massive solvent burdens. It requires the use of DMF in volumes up to 12.5 times the substrate weight, alongside ethyl acetate and isopropanol. DMF is notoriously difficult to remove due to its high boiling point and thermal stability, often leading to residual solvent issues in the final active pharmaceutical ingredient (API). Additionally, the use of methanol or isopropanol during the acidic deprotection step leads to the formation of ether-linked impurities, specifically identified as compounds of Formula-7, Formula-8, and Formula-9. These side reactions compromise the purity profile, forcing manufacturers to implement complex recrystallization sequences that further erode overall yield and increase cost reduction in pharmaceutical intermediates manufacturing efforts.
The Novel Approach
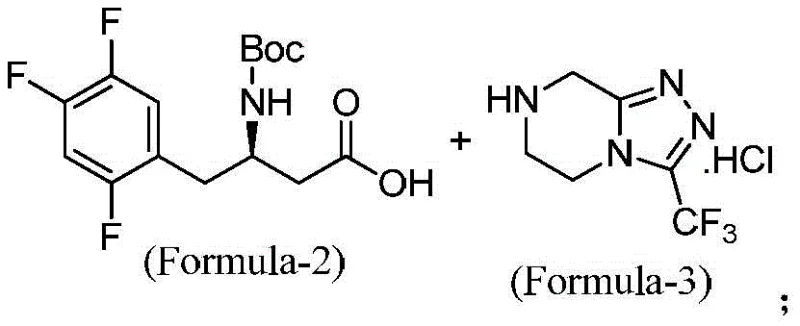
The innovative route presented in CN113149991A fundamentally reengineers the synthesis to overcome these specific deficits. By substituting dried HOBt with HOBt hydrate, the process eliminates the explosion risk while maintaining efficient coupling activity. More critically, the solvent system is optimized to exclude DMF entirely, favoring dichloromethane (DCM) which is far easier to recover and recycle via standard distillation techniques. The reaction sequence is designed as an in-situ process where the intermediate compound of Formula-4 is not isolated but directly subjected to deprotection conditions. This telescoping of steps not only saves time but also prevents the degradation of the intermediate that can occur during isolation and drying.
Furthermore, the novel approach strategically avoids the use of alcoholic solvents during the critical deprotection phase. By utilizing concentrated hydrochloric acid in a non-nucleophilic environment or carefully controlled conditions, the formation of the aforementioned ether impurities (Formula-7, 8, and 9) is effectively suppressed. The final crystallization employs methyl tert-butyl ether (MTBE) as an anti-solvent, which promotes the formation of high-purity crystals with a narrow particle size distribution. This results in a sitagliptin free base with an HPLC purity exceeding 99%, achieved through simple filtration rather than complex chromatography. For a commercial scale-up of complex pharmaceutical intermediates, this level of simplicity is invaluable, ensuring consistent quality and reliable supply continuity.
Mechanistic Insights into EDC-Mediated Amide Coupling and Deprotection
The chemical elegance of this synthesis lies in the precise control of the amide bond formation and the subsequent removal of the tert-butoxycarbonyl (Boc) protecting group. The coupling reaction utilizes N-(3-dimethylaminopropyl)-N'-ethylcarbodiimide hydrochloride (EDC.HCl) as the activating agent in the presence of HOBt hydrate. Mechanistically, EDC activates the carboxylic acid of the trifluorophenyl butanoic acid derivative (Formula-2) to form an O-acylisourea intermediate. This reactive species is rapidly intercepted by HOBt to generate a more stable and less racemization-prone active ester. The nucleophilic attack by the amine nitrogen of the triazolopyrazine hydrochloride (Formula-3) on this active ester drives the formation of the amide bond, yielding the protected intermediate (Formula-4). The use of HOBt hydrate is particularly noteworthy; the water of crystallization helps moderate the reactivity, preventing the formation of N-acylurea byproducts which are common dead-end impurities in carbodiimide couplings.
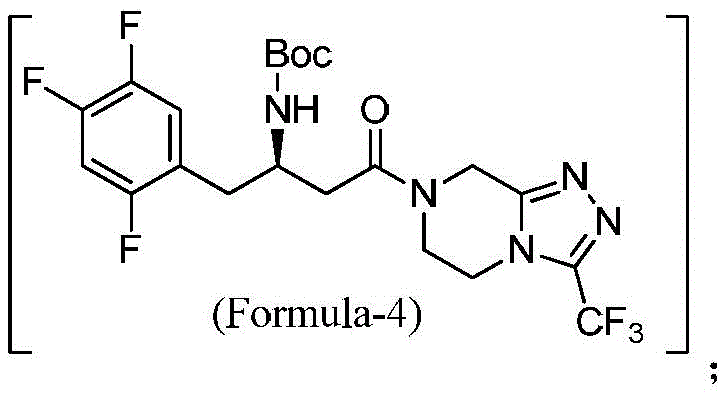
Following the coupling, the process moves to the deprotection phase, which is critical for defining the final impurity profile. In traditional acidic deprotections using alcohols like methanol or isopropanol, the carbocation generated from the Boc group cleavage can react with the alcohol solvent to form ether adducts on the sensitive triazolopyrazine ring or the phenyl backbone. The patented method avoids this by conducting the deprotection in a manner that minimizes nucleophilic competition from the solvent. By adjusting the pH to 10-12 post-reaction using sodium hydroxide, the free amine is liberated from its hydrochloride salt form. This basification step is crucial for extracting the neutral free base into the organic phase, leaving behind inorganic salts and water-soluble byproducts. The final vacuum distillation removes the extraction solvent, and the addition of MTBE induces crystallization. This mechanistic control ensures that the final product is free from the ether-linked impurities that plague older methods, securing a superior quality profile for downstream salt formation.
How to Synthesize Sitagliptin Free Base Efficiently
Implementing this synthesis requires strict adherence to temperature controls and stoichiometric balances to maximize the benefits of the in-situ design. The process begins with the activation of the acid component at low temperatures (0-5°C) to minimize side reactions, followed by a controlled warm-up to ambient conditions to drive the coupling to completion. The subsequent deprotection and workup phases rely on efficient phase separation and pH management to ensure high recovery. For detailed operational parameters, including specific stirring rates and addition profiles, operators should refer to the standardized technical documentation provided below.
- Couple (R)-3-(tert-butoxycarbonylamino)-4-(2,4,5-trifluorophenyl)butanoic acid with the triazolopyrazine hydrochloride using EDC.HCl and HOBt hydrate in dichloromethane at 0-30°C.
- Perform an aqueous workup to separate layers, followed by in-situ deprotection of the Boc-group using concentrated hydrochloric acid without isolating the intermediate carbamate.
- Adjust pH to 10-12 with alkali, extract into organic solvent, concentrate, and crystallize the final free base using methyl tert-butyl ether (MTBE) as an anti-solvent.
Commercial Advantages for Procurement and Supply Chain Teams
From a commercial perspective, the adoption of this synthesis route offers profound advantages for procurement managers and supply chain heads tasked with optimizing the cost of goods sold (COGS) and ensuring supply security. The most immediate impact is seen in the drastic simplification of the downstream processing train. By eliminating the need for column chromatography, the facility throughput is significantly increased, as batch cycles are shortened and equipment occupancy time is reduced. This operational efficiency directly correlates to lower manufacturing costs, as the expensive silica gel and large volumes of elution solvents associated with chromatography are completely removed from the bill of materials. Furthermore, the avoidance of DMF simplifies solvent recovery systems; DCM and MTBE have lower boiling points and form distinct azeotropes with water, making them energy-efficient to distill and recycle compared to the high-energy demand of stripping DMF.
Supply chain reliability is further enhanced by the improved safety profile of the reagents. The substitution of explosive anhydrous HOBt with the stable HOBt hydrate reduces the regulatory burden and insurance costs associated with storing energetic materials. This change also mitigates the risk of production stoppages due to safety incidents or stringent transport restrictions on hazardous chemicals. Additionally, the high purity of the crude product (>99% HPLC purity) means that fewer recrystallization cycles are needed to meet pharmacopeial standards. This consistency in quality reduces the variance in production timelines, allowing for more accurate forecasting and reducing lead time for high-purity pharmaceutical intermediates. The robustness of the process against impurity formation ensures that even at larger scales, the yield remains high and predictable, safeguarding against supply shortages.
- Cost Reduction in Manufacturing: The elimination of chromatographic purification represents a massive saving in both material and labor costs. Chromatography is notoriously expensive due to the cost of stationary phases and the vast quantities of solvents required for elution. By achieving high purity through crystallization alone, the process removes this cost center entirely. Additionally, the switch to easily recoverable solvents like DCM and MTBE lowers the net solvent consumption per kilogram of product, as recovery rates are higher and energy costs for distillation are lower compared to DMF-based systems.
- Enhanced Supply Chain Reliability: The use of commodity chemicals such as EDC.HCl, HOBt hydrate, and standard mineral acids ensures that the supply chain is not dependent on exotic or single-source reagents. The stability of HOBt hydrate allows for bulk purchasing and long-term storage without the degradation risks associated with anhydrous forms. This raw material security, combined with a process that is less sensitive to minor fluctuations in reaction conditions, ensures a steady and uninterrupted flow of product to downstream customers, minimizing the risk of stockouts.
- Scalability and Environmental Compliance: The process is inherently scalable because it relies on standard unit operations: reaction, extraction, distillation, and crystallization. There are no specialized steps that behave unpredictably upon scale-up. From an environmental standpoint, the reduction in solvent volume and the exclusion of DMF (a reproductive toxin with strict exposure limits) significantly lowers the facility's environmental footprint. The waste streams are easier to treat, and the lower E-factor (mass of waste per mass of product) aligns with modern green chemistry principles, facilitating smoother regulatory approvals and community relations.
Frequently Asked Questions (FAQ)
The following questions address common technical inquiries regarding the implementation and benefits of this specific sitagliptin synthesis pathway. These answers are derived directly from the experimental data and comparative analysis provided in the patent literature, offering clarity on how this method outperforms legacy technologies in terms of safety, purity, and efficiency.
Q: How does this process improve safety compared to traditional HOBt methods?
A: The patented method utilizes 1-hydroxybenzotriazole (HOBt) hydrate instead of explosive anhydrous HOBt. This substitution significantly mitigates the risk of detonation during handling and storage, enhancing overall plant safety protocols.
Q: What specific impurities are avoided by replacing methanol and IPA?
A: By eliminating methanol and isopropanol (IPA) from the deprotection and crystallization steps, the formation of ether-based impurities (specifically identified as Formula-7, Formula-8, and Formula-9 in the patent background) is effectively suppressed, leading to HPLC purity exceeding 99%.
Q: Why is the removal of DMF critical for commercial scalability?
A: Dimethylformamide (DMF) has a high boiling point and is difficult to remove completely, often requiring extensive washing with large volumes of other solvents. Replacing it with dichloromethane and MTBE allows for easier solvent recovery via distillation and reduces the environmental footprint of the waste stream.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Sitagliptin Free Base Supplier
At NINGBO INNO PHARMCHEM, we understand that the transition from laboratory scale to commercial production requires a partner with deep technical expertise and robust infrastructure. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the promising metrics seen in patent examples are realized in actual manufacturing campaigns. We maintain stringent purity specifications and operate rigorous QC labs equipped with state-of-the-art analytical instrumentation to verify that every batch of sitagliptin intermediate meets the highest global standards. Our commitment to quality assurance means that we can consistently deliver material that facilitates smooth downstream processing for our clients.
We invite pharmaceutical companies and contract manufacturers to collaborate with us to leverage this advanced synthesis technology. By partnering with us, you gain access to a Customized Cost-Saving Analysis tailored to your specific volume requirements and logistical needs. We encourage you to contact our technical procurement team today to request specific COA data and route feasibility assessments. Let us help you optimize your supply chain for sitagliptin production, ensuring cost-efficiency, safety, and uninterrupted supply for your critical diabetes medication portfolios.