Advanced Synthesis of 7-Benzyloxy-6-Methoxy-4-Hydroxyquinoline for Pharmaceutical Scale-Up
The pharmaceutical industry continuously demands more efficient and scalable routes for critical heterocyclic intermediates, particularly those serving as backbones for antimalarial and antitumor agents. Patent CN102030705B discloses a groundbreaking synthesis method for 7-benzyloxy-6-methoxy-4-hydroxyquinoline (Compound D), a pivotal scaffold in medicinal chemistry. This technology addresses the longstanding challenges of low yields and complex purification associated with traditional quinoline synthesis. By leveraging a novel one-pot reductive cyclization strategy, the process achieves high conversion rates and exceptional purity without the need for expensive chromatographic separation. As a reliable pharmaceutical intermediate supplier, understanding such technological leaps is crucial for securing supply chains for next-generation therapeutics. The following analysis details how this method transforms the manufacturing landscape for high-purity quinoline derivatives.
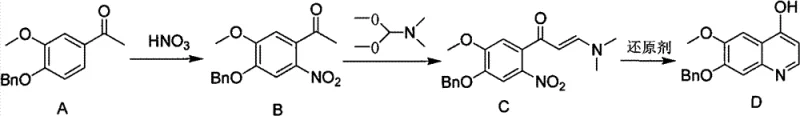
The structural integrity of Compound D, featuring a benzyloxy and methoxy substitution pattern on the quinoline ring, is essential for its biological activity in downstream drug applications. The patent highlights that this specific substitution pattern is notoriously difficult to install efficiently using classical methods. Conventional approaches often suffer from poor regioselectivity during the ring-closing step or require harsh conditions that degrade sensitive functional groups. The disclosed method overcomes these hurdles by utilizing a carefully orchestrated sequence of nitration, enamine formation, and finally, a mild reductive cyclization. This approach not only preserves the delicate ether linkages but also ensures the hydroxyl group at the 4-position is generated with high fidelity, setting a new standard for quality in API intermediate production.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Prior art technologies, such as those described in US6809097B1 and WO2006/108059, present significant bottlenecks for commercial manufacturing. The method in US6809097B1 relies on high-temperature heating to effect ring closure, which poses safety risks and increases energy consumption substantially. Furthermore, it necessitates silica gel column chromatography for purification, a technique that is prohibitively expensive and impractical for multi-kilogram or ton-scale production due to massive solvent usage and low throughput. Similarly, the process outlined in WO2006/108059 involves an excessive number of synthetic steps, leading to cumulative yield losses. With reported yields hovering between only 36% and 40%, this route results in substantial material waste and inflated production costs, making it economically unviable for cost-sensitive pharmaceutical projects. These limitations underscore the urgent need for a more robust and atom-economical synthetic pathway.
The Novel Approach
The methodology presented in CN102030705B represents a paradigm shift by consolidating the reduction and cyclization events into a single operational step. Instead of isolating unstable amine intermediates, the process generates the amine in situ via the reduction of a nitro group, which immediately undergoes cyclization to form the quinoline core. This telescoping of reactions minimizes handling losses and exposure of reactive intermediates to degradation. The use of inexpensive reducing agents like iron powder, coupled with common protonic acids such as acetic acid, drastically lowers the raw material costs. Moreover, the workup procedure is simplified to filtration and crystallization, completely eliminating the need for column chromatography. This streamlined workflow not only enhances safety by avoiding high-temperature operations but also significantly improves the overall process mass intensity, aligning perfectly with green chemistry principles and modern manufacturing efficiency goals.
Mechanistic Insights into Reductive Cyclization and Nitration
The success of this synthesis hinges on the precise control of three distinct chemical transformations, beginning with the regioselective nitration of the acetophenone precursor. As illustrated in the reaction scheme, Compound A undergoes nitration using nitric acid in a polar solvent system, which may include acetic anhydride or sulfuric acid. The reaction is conducted at low temperatures, typically between -5°C and 10°C, to prevent over-nitration and oxidative degradation of the electron-rich aromatic ring. This step installs the critical nitro group ortho to the acetyl moiety, setting the stage for the subsequent ring closure. The choice of solvent and acid concentration is vital here; the patent notes that diluting concentrated nitric acid with a polar solvent helps reduce by-product formation, thereby enhancing the purity of Compound B before it proceeds to the next stage.
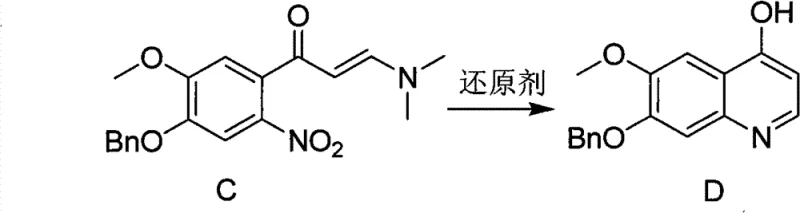
Following nitration, the ketone functionality is converted into an enamine intermediate (Compound C) through reaction with N,N-dimethylformamide dimethyl acetal (DMF-DMA). This transformation activates the alpha-carbon for nucleophilic attack during the cyclization phase. The final and most critical step is the reductive cyclization depicted in the figure above. Under the influence of a protonic acid and a reducing agent like iron powder, the nitro group of Compound C is reduced to an amine. This newly formed amine then attacks the activated enamine carbon, closing the ring to form the quinoline system. The reaction is typically run at moderate temperatures of 50°C to 100°C in protic solvents like ethanol or acetic acid. This mechanism ensures high selectivity for the 4-hydroxyquinoline product, minimizing the formation of regioisomers or polymeric impurities that often plague quinoline syntheses. The result is a crude product with purity exceeding 95%, which can be further upgraded to pharmaceutical grade through simple recrystallization.
How to Synthesize 7-Benzyloxy-6-Methoxy-4-Hydroxyquinoline Efficiently
Implementing this synthesis requires strict adherence to the optimized conditions regarding stoichiometry, temperature, and solvent selection to maximize yield and safety. The process begins with the preparation of the nitro-acetophenone intermediate, followed by enamine formation, and concludes with the tandem reduction-cyclization. Each step has been fine-tuned to balance reaction rate with impurity control, ensuring that the final isolation is straightforward. For R&D teams looking to replicate or scale this process, the detailed standardized synthetic steps provided below offer a robust framework for technology transfer. These guidelines reflect the optimal parameters identified in the patent examples, ensuring reproducible results from the laboratory bench to the pilot plant.
- Perform regioselective nitration of 4-benzyloxy-5-methoxyacetophenone (Compound A) using diluted nitric acid in a polar solvent at low temperatures (-5 to 10°C) to obtain Compound B.
- React Compound B with N,N-dimethylformamide dimethyl acetal (DMF-DMA) in a polar aprotic solvent at 80-120°C to form the enamine intermediate, Compound C.
- Execute a one-pot reductive cyclization of Compound C using iron powder and a protonic acid (e.g., acetic acid) at 50-100°C, followed by crystallization to isolate Compound D.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this synthesis method offers tangible strategic benefits beyond mere technical elegance. The primary advantage lies in the drastic simplification of the downstream processing workflow. By removing the requirement for silica gel column chromatography, manufacturers can eliminate a major bottleneck that typically limits batch size and extends production lead times. This change alone translates into significant cost reduction in pharmaceutical intermediate manufacturing, as it reduces solvent consumption, waste disposal costs, and labor hours associated with purification. Furthermore, the use of commodity chemicals like iron powder and acetic acid instead of precious metal catalysts or exotic reagents stabilizes the raw material supply chain against market volatility. This resilience is critical for maintaining continuous production schedules and meeting tight delivery windows for global clients.
- Cost Reduction in Manufacturing: The elimination of chromatographic purification and the use of inexpensive iron powder as a reductor fundamentally alter the cost structure of production. Traditional methods relying on column separation incur high operational expenses due to solvent recovery and silica disposal. In contrast, this method utilizes crystallization, a unit operation that is inherently scalable and cost-effective. Additionally, the high yield of the reductive cyclization step (reported greater than 80%) means less starting material is wasted, directly improving the cost of goods sold (COGS). The overall three-step sequence achieves a total yield of 54% to 77%, which is a substantial improvement over the sub-40% yields of legacy processes, effectively doubling the output per unit of input material.
- Enhanced Supply Chain Reliability: The reliance on readily available starting materials and common solvents ensures that production is not held hostage by the availability of specialized reagents. Compound A and the necessary reagents like DMF-DMA and nitric acid are bulk commodities with stable global supply networks. This accessibility reduces the risk of supply disruptions that can occur with niche catalysts or custom-synthesized building blocks. Moreover, the mild reaction conditions (50-100°C) reduce the stress on reactor equipment and lower the energy burden compared to high-temperature alternatives. This operational robustness allows for more predictable manufacturing cycles, enabling supply chain planners to commit to firmer delivery dates and maintain healthier inventory levels without the fear of batch failures.
- Scalability and Environmental Compliance: From an environmental and regulatory standpoint, this process aligns well with modern green chemistry mandates. The atom economy is superior due to the direct cyclization mechanism, and the waste stream is significantly cleaner, consisting mainly of inorganic salts from the iron reduction which are easier to treat than organic solvent sludge from chromatography. The ability to scale this process from 100 kgs to 100 MT annual commercial production is facilitated by the simplicity of the workup—filtration and washing are easily automated in large-scale filter dryers. This scalability ensures that as demand for the downstream API grows, the supply of this critical intermediate can be ramped up rapidly without requiring complex process re-engineering or new capital-intensive purification infrastructure.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis route. These insights are derived directly from the experimental data and comparative analysis provided in the patent documentation. Understanding these nuances helps stakeholders evaluate the feasibility of integrating this technology into their existing manufacturing portfolios. The answers reflect the specific advantages in purity, yield, and operational safety that distinguish this method from conventional alternatives.
Q: What are the key advantages of this synthesis method over prior art?
A: This method eliminates the need for high-temperature ring closure and silica gel column chromatography required in previous patents (e.g., US6809097B1). It achieves higher yields (>80% vs 36-40%) and uses simpler post-processing like filtration and crystallization, making it ideal for industrial scale-up.
Q: What reducing agents are suitable for the cyclization step?
A: The patent specifies that iron powder and/or zinc powder can be used, with iron powder being the preferred choice due to its cost-effectiveness and high efficiency in the presence of protonic acids like acetic acid or hydrochloric acid.
Q: Is this process suitable for large-scale manufacturing?
A: Yes, the process is explicitly designed for industrial production. It avoids hazardous high-temperature conditions and complex purification steps, utilizing commercially available raw materials and standard solvents like ethanol and acetic acid.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 7-Benzyloxy-6-Methoxy-4-Hydroxyquinoline Supplier
The synthesis of 7-benzyloxy-6-methoxy-4-hydroxyquinoline described in CN102030705B represents a significant opportunity for optimizing the supply of antimalarial and anticancer drug precursors. At NINGBO INNO PHARMCHEM, we possess the technical expertise to translate such patented methodologies into robust commercial processes. Our team has extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the theoretical benefits of high yield and purity are realized in actual manufacturing. We operate stringent purity specifications and utilize rigorous QC labs to guarantee that every batch of intermediate meets the exacting standards required by global regulatory bodies. Our commitment to quality assurance means that we can deliver high-purity pharmaceutical intermediates that facilitate smoother downstream synthesis for our partners.
We invite pharmaceutical companies and research institutions to collaborate with us to leverage this advanced synthesis technology. By partnering with us, you gain access to a Customized Cost-Saving Analysis tailored to your specific volume requirements and quality targets. We encourage you to contact our technical procurement team to request specific COA data and route feasibility assessments. Whether you require small quantities for clinical trials or metric tons for commercial launch, our flexible manufacturing capabilities and deep process knowledge position us as the ideal partner for securing your supply chain of complex quinoline intermediates.