Advanced Synthesis of High-Purity Fluticasone Furoate for Scalable Pharmaceutical Manufacturing
The pharmaceutical industry continuously demands higher purity standards for corticosteroid intermediates, particularly for potent anti-inflammatory agents like fluticasone furoate. Patent CN109438543B introduces a groundbreaking preparation method that addresses the longstanding challenges of impurity control and yield optimization in the synthesis of this critical active pharmaceutical ingredient (API). The core innovation lies in a refined synthetic pathway that transforms a thiocarbamate precursor (Formula III) directly into the target S-fluoromethyl ester (Formula I) via a stabilized intermediate solution, effectively bypassing the degradation pitfalls of traditional isolation techniques. This technical breakthrough not only enhances the chemical integrity of the final product but also streamlines the manufacturing workflow, offering a compelling value proposition for global supply chains seeking reliable sources of high-quality respiratory medication components.
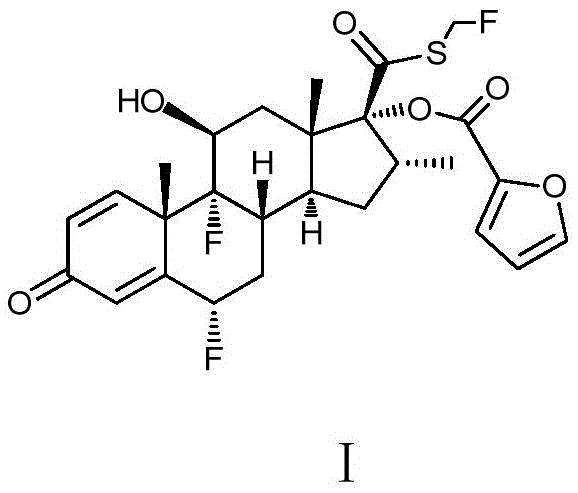
Fluticasone furoate, chemically defined as 6 alpha, 9 alpha-difluoro-17 alpha-[(2-furyl carbonyl)oxy]-11 beta-hydroxy-16 alpha-methyl-3-oxo-androstane-1, 4-diene-17 beta-thiocarboxylic acid-S-fluoromethyl ester, serves as a vital therapeutic agent for asthma and allergic rhinitis. The structural complexity of this molecule, particularly the sensitive 17-beta position, necessitates precise synthetic control to prevent the formation of deleterious by-products such as methyl esters or oxidation derivatives. The patented methodology leverages a strategic alcoholysis followed by a nucleophilic substitution, ensuring that the reactive thiol intermediate remains protected within a optimized solvent matrix until the final fluoromethylation step. This approach represents a significant paradigm shift from legacy processes, positioning manufacturers to meet stringent regulatory requirements for related substances while maintaining economic viability in a competitive market.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
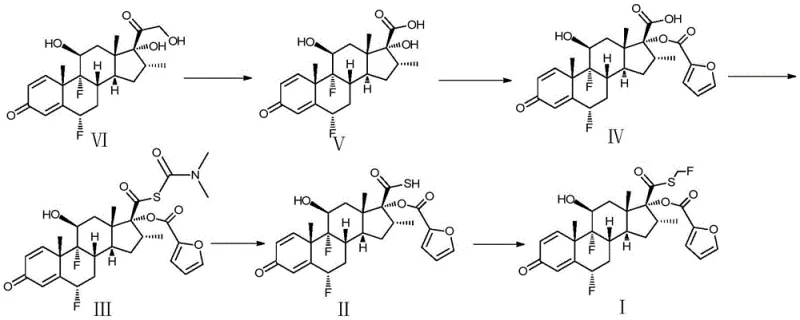
Historically, the synthesis of fluticasone furoate has been plagued by the inherent instability of the key thiol intermediate (Formula II). Prior art methods, such as those disclosed in earlier patents, often required the isolation of this intermediate as a solid or as an unstable organic amine salt. This isolation step proved to be a critical bottleneck; the solid Formula II is highly susceptible to oxidation and difficult to filter, leading to significant material loss and yield reductions often ranging between 10% to 20% for the overall sequence. Furthermore, the purification of Formula II via salt formation introduced additional complexity, requiring multiple acid-base cycles that failed to effectively remove core impurities like methyl esters. The reliance on hazardous reagents like hydrogen sulfide in some precursor syntheses further compounded safety risks, making these conventional routes less attractive for modern, safety-conscious industrial facilities aiming for green chemistry compliance.
The Novel Approach
In stark contrast to these cumbersome legacy pathways, the novel approach detailed in CN109438543B initiates the final sequence from the stable thiocarbamate precursor (Formula III). By employing a direct alcoholysis reaction in a controlled alcoholic medium, the process generates the reactive thiol (Formula II) in situ, maintaining it within a solution phase that minimizes its exposure to oxidative conditions. This "telescoped" strategy eliminates the need to isolate, filter, and dry the unstable intermediate, thereby drastically reducing the residence time of Formula II in a vulnerable state. The subsequent substitution reaction utilizes this purified solution directly, reacting it with a fluorohalomethylation reagent to forge the critical S-C bond. This seamless integration of reaction and workup steps not only simplifies the operational procedure but also significantly boosts the cumulative yield of the final two steps to over 80%, demonstrating a clear superiority in both efficiency and product quality.
Mechanistic Insights into Alcoholysis and Fluoromethylation
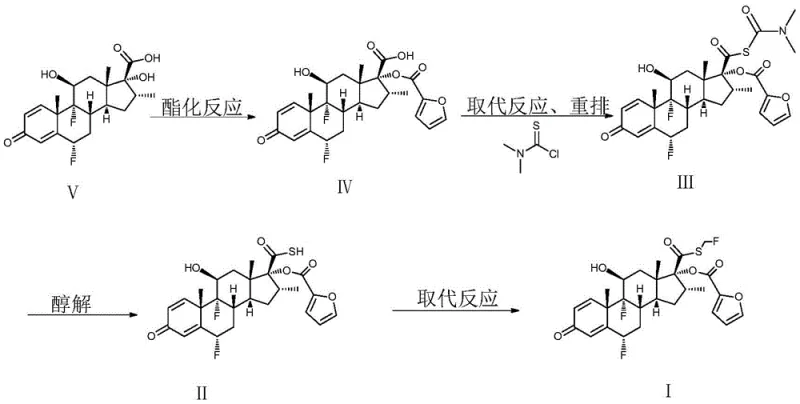
The chemical elegance of this process is rooted in the precise management of the alcoholysis mechanism. When Formula III is treated with an inorganic base, such as potassium carbonate, in a lower alcohol solvent like methanol, the thiocarbamate moiety undergoes cleavage to release the free thiol (Formula II). However, this reaction concurrently generates fat-soluble impurities and potentially unreacted starting material. The innovation here is not merely the reaction itself, but the sophisticated post-treatment protocol designed to purify this mixture without precipitation. By utilizing a specific biphasic or triphasic solvent system comprising water, alcohols, and carefully selected organic solvents like methyl tert-butyl ether (MTBE) and toluene, the process achieves a highly selective partitioning of impurities. The organic phases extract lipophilic contaminants, while the aqueous phase retains the ionized or polar species, allowing for the recovery of a highly enriched solution of Formula II ready for the next transformation.
Following purification, the substitution mechanism involves the nucleophilic attack of the thiol sulfur on the fluorohalomethane reagent, facilitated by an organic base like N,N-diisopropylethylamine. This step is critical for installing the S-fluoromethyl group, which is essential for the biological activity of the drug. The reaction conditions are meticulously controlled at low temperatures (-10°C to 0°C) to suppress side reactions and ensure regioselectivity. The final recrystallization step employs a ketone solvent mixed with a normal alkane, such as n-heptane, to induce crystallization. This specific solvent pairing is engineered to maximize the exclusion of trace impurities, including any residual methyl esters or oxidation products, ensuring that the final crystal lattice of fluticasone furoate achieves the exceptional purity levels required for inhalation therapies, often exceeding 99.8%.
How to Synthesize Fluticasone Furoate Efficiently
The implementation of this advanced synthesis route requires strict adherence to the optimized parameters regarding solvent ratios, temperature controls, and pH adjustments to ensure reproducibility and high yield. The process is designed to be robust, allowing for the direct use of the crude intermediate solution in the subsequent substitution step, which is a key factor in its commercial viability. For R&D teams and process engineers looking to adopt this methodology, the following guide outlines the critical operational phases derived from the patent examples, emphasizing the importance of the extraction workup in maintaining intermediate stability.
- Perform alcoholysis on compound III using an alcohol solvent and alkali to generate a mixture containing compound II without isolation.
- Execute a multi-stage liquid-liquid extraction workup to remove fat-soluble impurities and unreacted starting materials from the compound II mixture.
- Conduct a substitution reaction on the purified compound II solution with a fluorohalomethylation reagent, followed by recrystallization to obtain high-purity compound I.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this novel synthesis route offers tangible strategic benefits that extend beyond simple chemical yield. By fundamentally altering the process flow to eliminate the isolation of the unstable intermediate, manufacturers can achieve significant cost reduction in API manufacturing through the reduction of unit operations. The removal of filtration, drying, and re-dissolution steps for Formula II translates directly into lower energy consumption, reduced solvent usage, and decreased labor hours per batch. Furthermore, the avoidance of hazardous reagents like hydrogen sulfide simplifies waste treatment protocols and lowers the environmental compliance burden, contributing to a more sustainable and cost-effective production model that aligns with modern ESG (Environmental, Social, and Governance) goals.
- Cost Reduction in Manufacturing: The streamlined process significantly lowers the cost of goods sold (COGS) by improving the overall material throughput. By achieving a combined yield of over 80% for the final conversion steps compared to the much lower yields of prior art, the effective cost per kilogram of the active ingredient is drastically reduced. Additionally, the simplified workup reduces the demand for specialized equipment and minimizes solvent recovery costs, providing a clear economic advantage for large-scale production runs without compromising on the stringent quality specifications required for respiratory drugs.
- Enhanced Supply Chain Reliability: The robustness of this synthetic route enhances supply chain continuity by reducing the risk of batch failures associated with the difficult purification of unstable intermediates. Traditional methods often suffered from low yields and inconsistent purity due to the sensitivity of Formula II, leading to potential supply disruptions. This new method's ability to consistently produce high-purity material with minimal impurities ensures a more predictable manufacturing schedule, allowing suppliers to meet tight delivery windows and maintain steady inventory levels for downstream formulation partners.
- Scalability and Environmental Compliance: Designed with industrial amplification in mind, this process avoids the use of highly toxic chemicals and complex salt-forming purification steps that are difficult to scale safely. The use of common, recyclable solvents like MTBE, toluene, and heptane facilitates easier solvent recovery and waste management. This scalability ensures that the transition from pilot plant to commercial tonnage production is smooth and efficient, enabling suppliers to rapidly ramp up capacity to meet global market demand for fluticasone furoate while adhering to strict environmental regulations.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production of fluticasone furoate using this advanced methodology. These insights are derived directly from the experimental data and comparative analysis provided in the patent documentation, offering clarity on how this process outperforms historical benchmarks in terms of purity, safety, and operational efficiency.
Q: How does this new method improve the purity of Fluticasone Furoate compared to prior art?
A: The novel process avoids isolating the unstable intermediate (Formula II), which is prone to oxidation and methyl ester formation in traditional methods. By keeping Formula II in solution and utilizing a specific multi-solvent extraction system, fat-soluble impurities are effectively removed, achieving purities exceeding 99.8%.
Q: What are the safety advantages of this synthesis route for industrial scale-up?
A: Unlike some prior art methods that require hazardous hydrogen sulfide or similar compounds for starting material synthesis, this route utilizes safer reagents like fluorobromomethane and standard inorganic bases. Furthermore, eliminating the filtration and drying of the unstable intermediate reduces operational risks and potential exposure to reactive species.
Q: Is this process suitable for large-scale commercial production?
A: Yes, the method is explicitly designed for industrial amplification. It simplifies the workflow by removing complex salt-formation purification steps and difficult filtration procedures associated with intermediate Formula II, resulting in a robust process with a combined yield of over 80% for the final two steps.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Fluticasone Furoate Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of purity and consistency in the supply of corticosteroid intermediates for the global pharmaceutical market. As a dedicated CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that our clients receive material that meets the highest industry standards. Our state-of-the-art facilities are equipped with rigorous QC labs capable of verifying stringent purity specifications, including the precise control of related substances and residual solvents, guaranteeing that every batch of fluticasone furoate we deliver is ready for immediate formulation into life-saving inhalation therapies.
We invite forward-thinking pharmaceutical companies to collaborate with us to leverage this advanced synthesis technology for their supply chains. By partnering with our technical procurement team, you can access a Customized Cost-Saving Analysis tailored to your specific volume requirements, demonstrating how our optimized process can reduce your overall manufacturing expenses. We encourage you to contact us today to request specific COA data and route feasibility assessments, ensuring that your project moves forward with a reliable, high-quality, and cost-effective supply of this essential API intermediate.