Scalable Synthesis of 5-Fluoro-isoindole-1,3-dione Derivatives for Advanced Oncology Intermediates
Scalable Synthesis of 5-Fluoro-isoindole-1,3-dione Derivatives for Advanced Oncology Intermediates
The pharmaceutical landscape is increasingly driven by the demand for specialized intermediates capable of supporting next-generation therapeutic modalities, particularly Proteolysis Targeting Chimeras (PROTACs) and immunomodulatory imide drugs (IMiDs). Patent CN114773316A introduces a robust and highly efficient preparation method for 2-(2,6-dioxo-piperidin-3-yl)-5-fluoro-isoindole-1,3-dione, a critical building block in the synthesis of fluorinated thalidomide analogues. This technical disclosure addresses the longstanding challenges associated with the industrial production of such complex heterocyclic compounds, specifically focusing on yield optimization and purity enhancement. By leveraging a strategic halogen-magnesium exchange followed by precise carboxylation and cyclization steps, the patented process offers a viable pathway for manufacturing high-value oncology intermediates. For R&D directors and procurement specialists, understanding the nuances of this synthetic route is essential for securing a reliable supply chain for advanced drug development programs.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of fluorinated isoindole-1,3-dione derivatives has been plagued by inefficiencies that hinder large-scale commercialization. Traditional routes often rely on multi-step sequences involving harsh reaction conditions, expensive transition metal catalysts, or precursors that are difficult to source in bulk quantities. These conventional methodologies frequently suffer from poor atom economy and generate significant amounts of hazardous waste, complicating environmental compliance and driving up production costs. Furthermore, achieving the stringent purity profiles required for clinical-grade intermediates often necessitates extensive purification protocols, such as repeated chromatography, which are impractical for ton-scale manufacturing. The cumulative effect of these limitations is a market characterized by high prices and inconsistent supply availability, posing significant risks to pharmaceutical developers aiming to bring novel anti-tumor agents to market efficiently.
The Novel Approach
The methodology outlined in the patent represents a paradigm shift by utilizing 2-bromo-4-fluorobenzoic acid as a cost-effective starting material. This approach streamlines the synthesis into three distinct, high-yielding stages that avoid the need for exotic reagents. The core innovation lies in the controlled generation of a Grignard species followed by immediate carboxylation to install the second carboxylic acid functionality, effectively constructing the phthalic acid backbone in situ. Subsequent dehydration and condensation steps are optimized to proceed under relatively mild thermal conditions, ensuring the integrity of the sensitive fluorine substituent. This strategic design not only simplifies the operational workflow but also inherently reduces the formation of difficult-to-remove impurities, thereby facilitating a more direct route to the final high-purity product suitable for downstream drug synthesis.
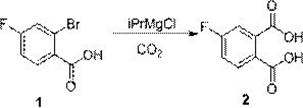
Mechanistic Insights into Grignard Carboxylation and Cyclization
The initial step of this synthesis involves a delicate halogen-magnesium exchange reaction, which serves as the foundation for the entire molecular architecture. In this phase, 2-bromo-4-fluorobenzoic acid reacts with isopropyl magnesium chloride (iPrMgCl) in tetrahydrofuran (THF) to generate the corresponding aryl magnesium species. The patent emphasizes the critical importance of temperature control during this exothermic process, specifying a range of 20-30°C to ensure complete conversion while minimizing side reactions such as Wurtz-type coupling. Following the exchange, the reaction mixture is cooled to 0-10°C before the introduction of carbon dioxide gas. This thermal management is vital because the subsequent quenching with CO2 is highly exothermic; maintaining a lower temperature prevents the degradation of the organometallic intermediate and ensures the selective formation of the dicarboxylic acid, 4-fluoro-1,2-phthalic acid, with high regioselectivity.
Following the formation of the dicarboxylic acid, the process transitions to a dehydration cyclization to form the anhydride, followed by a nucleophilic substitution. The dehydration is effected using acetic anhydride at elevated temperatures (around 105°C), which drives the equilibrium towards the cyclic anhydride by removing water. The final coupling with 3-amino-2,6-piperidinedione hydrochloride is conducted in acetic acid with sodium acetate acting as a base. This specific solvent system and base combination are crucial for solubilizing the reactants and facilitating the nucleophilic attack of the amine on the anhydride carbonyl. The mechanism proceeds through an amic acid intermediate which spontaneously cyclizes under the reaction conditions to form the stable isoindole-1,3-dione ring system. The careful selection of sodium acetate helps buffer the reaction environment, preventing the racemization of the chiral center on the piperidine ring and ensuring the final product achieves the reported purity of 99.7%.
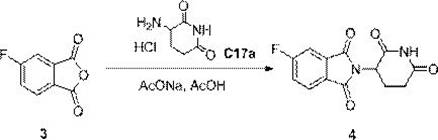
How to Synthesize 2-(2,6-dioxo-piperidin-3-yl)-5-fluoro-isoindole-1,3-dione Efficiently
Implementing this synthesis requires strict adherence to the optimized parameters regarding stoichiometry, temperature, and workup procedures to replicate the high yields reported in the patent examples. The process begins with the preparation of the Grignard reagent and its reaction with the bromo-acid, followed by a rigorous aqueous workup to isolate the phthalic acid intermediate. The subsequent dehydration and coupling steps demand precise thermal control to avoid decomposition of the sensitive anhydride or the amino-piperidine component. Operators must ensure that solvent volumes and drying times are sufficient to remove residual moisture, which could otherwise hydrolyze the anhydride or interfere with the final cyclization. For a detailed breakdown of the specific operational parameters, including exact molar ratios and stirring times, please refer to the standardized synthesis guide below.
- Perform halogen-magnesium exchange on 2-bromo-4-fluorobenzoic acid using iPrMgCl, followed by CO2 quenching to yield 4-fluoro-1,2-phthalic acid.
- Execute dehydration of the phthalic acid intermediate using acetic anhydride at elevated temperatures to form 4-fluorophthalic anhydride.
- Conduct nucleophilic substitution between the anhydride and 3-amino-2,6-piperidinedione in acetic acid with sodium acetate to finalize the product.
Commercial Advantages for Procurement and Supply Chain Teams
From a supply chain perspective, the adoption of this synthetic route offers compelling advantages related to raw material security and operational expenditure. The primary starting material, 2-bromo-4-fluorobenzoic acid, is a commodity chemical available from multiple global suppliers, which mitigates the risk of single-source dependency often associated with specialized fine chemicals. Furthermore, the elimination of precious metal catalysts, such as palladium or rhodium, which are common in alternative cross-coupling strategies, removes the need for expensive metal scavenging steps and reduces the burden of heavy metal testing in the final quality control phase. This simplification of the purification train translates directly into reduced processing time and lower utility consumption, making the overall manufacturing process more economically sustainable and environmentally friendly for large-scale production facilities.
- Cost Reduction in Manufacturing: The economic viability of this process is significantly enhanced by the use of inexpensive reagents like isopropyl magnesium chloride and acetic anhydride, which are produced on a massive industrial scale. By avoiding complex catalytic cycles and utilizing straightforward crystallization techniques for purification, the process minimizes the consumption of high-cost chromatographic media and solvents. The high yield observed in the final coupling step, approaching quantitative levels in some examples, ensures that the valuable chiral piperidine precursor is utilized with maximum efficiency, drastically reducing the cost of goods sold (COGS) per kilogram of the final intermediate.
- Enhanced Supply Chain Reliability: The robustness of the reaction conditions contributes to a more predictable manufacturing timeline. The tolerance of the Grignard step to slight variations in temperature and the use of common solvents like THF and acetic acid mean that the process can be easily transferred between different manufacturing sites without requiring specialized equipment. This flexibility allows for diversified production strategies, ensuring continuity of supply even in the face of regional disruptions. Additionally, the stability of the intermediates allows for potential campaign manufacturing, where the phthalic acid or anhydride can be stockpiled, further buffering the supply chain against demand fluctuations.
- Scalability and Environmental Compliance: The process is inherently designed for scale-up, utilizing reaction temperatures and pressures that are standard in existing chemical infrastructure. The waste profile is favorable, primarily consisting of magnesium salts and acetic acid byproducts, which are easier to treat and dispose of compared to the heavy metal waste streams generated by transition-metal catalyzed routes. This alignment with green chemistry principles facilitates smoother regulatory approvals and reduces the environmental compliance costs associated with waste management, positioning this method as a preferred choice for sustainable pharmaceutical manufacturing.
Frequently Asked Questions (FAQ)
The following questions address common technical inquiries regarding the implementation and optimization of this synthetic pathway. These insights are derived directly from the experimental data and embodiments disclosed in the patent documentation, providing clarity on critical process parameters. Understanding these details is crucial for process chemists aiming to adapt this technology for their specific production needs and for procurement teams evaluating the technical feasibility of the supply source.
Q: What are the critical reaction conditions for the Grignard step in this synthesis?
A: The patent specifies a temperature range of 20-30°C for the exchange reaction, with a critical cooling step to 0-10°C prior to CO2 quenching to maximize yield and minimize side reactions.
Q: How does this process improve upon conventional methods for fluorinated thalidomide analogues?
A: This route utilizes readily available 2-bromo-4-fluorobenzoic acid and avoids complex catalytic systems, resulting in a final product purity of up to 99.7% and significantly simplified downstream processing.
Q: Is this synthesis suitable for large-scale industrial production?
A: Yes, the use of common solvents like THF and acetic acid, along with mild reaction temperatures and robust workup procedures involving crystallization, indicates strong potential for commercial scale-up.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 2-(2,6-dioxo-piperidin-3-yl)-5-fluoro-isoindole-1,3-dione Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that high-quality intermediates play in the development of life-saving oncology therapies. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from laboratory bench to industrial reactor is seamless and efficient. We are committed to delivering products that meet stringent purity specifications, supported by our rigorous QC labs equipped with state-of-the-art analytical instrumentation. Our expertise in handling sensitive organometallic reactions and complex heterocyclic syntheses allows us to consistently reproduce the high yields and purity profiles described in leading patents, guaranteeing a supply of material that accelerates your drug development timelines.
We invite you to collaborate with us to optimize your supply chain for this vital intermediate. Our technical procurement team is ready to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements, demonstrating how our manufacturing efficiencies can translate into tangible value for your organization. Please contact us today to request specific COA data and route feasibility assessments, and let us demonstrate why we are the preferred partner for your complex pharmaceutical intermediate needs.