Advanced One-Step Synthesis of Teniposide Intermediate for Commercial Scale-up
Advanced One-Step Synthesis of Teniposide Intermediate for Commercial Scale-up
The pharmaceutical industry continuously seeks robust and scalable pathways for the production of potent antineoplastic agents, and Teniposide remains a cornerstone therapy for treating malignant lymphoma and acute lymphocytic leukemia. A pivotal breakthrough in this domain is documented in patent CN112079879B, which discloses a novel synthesis method for the critical intermediate, 4'-demethylepipodophyllotoxin-beta-D-glucopyranoside. This intellectual property addresses the longstanding bottlenecks associated with the deprotection of complex lignan glycosides, offering a streamlined alternative to legacy processes that rely on hazardous hydrogenation and expensive Lewis acids. For R&D directors and supply chain strategists, this technology represents a significant opportunity to enhance the reliability of the pharmaceutical intermediates supply chain while simultaneously driving down the cost of goods sold through process intensification.
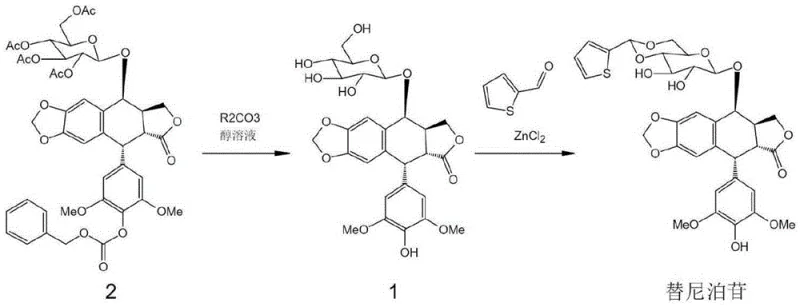
The core innovation lies in the ability to transform the fully protected precursor, Compound 2, directly into the bioactive-ready Intermediate 1 in a single operational step. Historically, the synthesis of this key building block has been plagued by low overall yields and complex purification requirements, which directly impact the commercial viability of the final API. By leveraging common and inexpensive alkali carbonates as catalysts in an alcoholic medium, this new methodology achieves selective deprotection under mild conditions. This approach not only preserves the sensitive stereochemistry of the podophyllotoxin scaffold but also eliminates the need for transition metal catalysts, aligning perfectly with modern green chemistry principles and stringent regulatory expectations for residual metal limits in oncology drugs.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Prior art methodologies for synthesizing the Teniposide intermediate typically involve a cumbersome two-step sequence that introduces significant inefficiencies into the manufacturing workflow. The traditional route first employs anhydrous zinc acetate in refluxing methanol for over 20 hours to remove acetyl protecting groups, a process that often yields less than 50% of the desired mono-deprotected species due to competing side reactions and incomplete conversion. Following this, a second distinct step utilizing palladium on carbon and hydrogen gas is required to cleave the benzyloxycarbonyl (Cbz) group, which adds further complexity and risk. The cumulative yield of this two-step sequence is frequently reported to be less than 40%, implying that more than half of the valuable starting material is lost to waste streams. Furthermore, the reliance on hydrogenation necessitates specialized high-pressure equipment and rigorous safety protocols, while the use of zinc salts generates heavy metal waste that requires costly disposal and extensive purification to meet pharmacopeial standards.
The Novel Approach
In stark contrast, the novel method described in the patent utilizes a simple yet highly effective one-pot strategy that collapses the deprotection sequence into a single unit operation. By dissolving Compound 2 in a lower alcohol such as methanol or ethanol and treating it with an alkali metal carbonate (where the metal can be lithium, sodium, potassium, rubidium, cesium, or francium), both the acetyl and Cbz protecting groups are removed simultaneously. This reaction proceeds efficiently at temperatures ranging from -5°C to 25°C, eliminating the energy-intensive reflux conditions of the past. The result is a dramatic improvement in process mass intensity, with isolated yields of Intermediate 1 reaching between 72% and 89% depending on the specific carbonate and temperature employed. This consolidation of steps not only doubles the effective throughput of the reactor but also simplifies the downstream processing, as the workup involves merely filtering off solids and crystallizing the product from an acetone-water mixture.
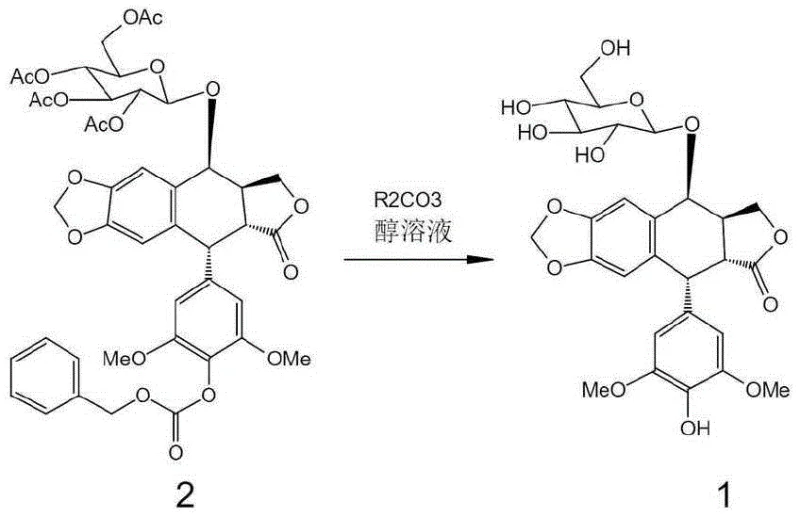
Mechanistic Insights into Alkali Carbonate Catalyzed Deprotection
The success of this synthesis hinges on the precise modulation of nucleophilicity and basicity provided by the alkali carbonate catalyst in an alcoholic solvent. Unlike strong bases such as sodium hydroxide, which would indiscriminately hydrolyze the sensitive lactone ring and potentially open the acetal linkage of the glycoside, alkali carbonates offer a buffered basic environment. This mild alkalinity is sufficient to catalyze the transesterification of the acetate groups on the sugar moiety and the aminolysis-like cleavage of the carbamate (Cbz) group, yet it remains below the threshold required to attack the sterically hindered and electronically stable lactone carbonyl of the podophyllotoxin core. The reaction mechanism likely involves the generation of alkoxide ions in situ, which act as the active nucleophiles attacking the ester and carbamate functionalities. The choice of solvent, specifically methanol or ethanol, plays a dual role as both the reaction medium and the source of the alkoxy nucleophile, ensuring that the deprotection proceeds via a clean transesterification pathway rather than hydrolysis, which minimizes the formation of acidic byproducts that could degrade the acid-sensitive acetal bridge.
From an impurity control perspective, this mechanistic pathway offers superior selectivity compared to Lewis acid-catalyzed methods. The traditional zinc acetate route operates under conditions where the Lewis acidity can promote the opening of the acetal ring or the epimerization of the chiral centers, leading to difficult-to-remove diastereomeric impurities. By avoiding acidic conditions entirely and maintaining a near-neutral to mildly basic pH throughout the reaction, the novel process preserves the optical purity of the molecule. Additionally, the absence of transition metals like zinc or palladium means that the final product is inherently free from heavy metal contamination, a critical quality attribute for oncology APIs. The purification strategy described, involving concentration and crystallization from acetone and water, leverages the solubility differences between the polar deprotected intermediate and any remaining non-polar impurities, further enhancing the chemical purity without the need for chromatographic separation.
How to Synthesize 4'-Demethylepipodophyllotoxin-beta-D-glucopyranoside Efficiently
The implementation of this synthesis route is straightforward and amenable to standard batch reactor configurations found in most multipurpose pharmaceutical facilities. The process begins with the dissolution of the starting material in methanol, followed by the controlled addition of the carbonate base at low temperatures to manage the exotherm and ensure selectivity. After a reaction period of approximately 16 hours, the mixture is filtered to remove insoluble carbonate salts, and the filtrate is neutralized with dilute acetic acid to quench any remaining base. The final isolation is achieved through a solvent swap to an acetone-water system, which induces crystallization of the high-purity intermediate. For detailed operational parameters, stoichiometry, and specific workup instructions, please refer to the standardized protocol below.
- Dissolve the fully protected precursor (Compound 2) in an alcoholic solvent such as methanol or ethanol, optionally applying slight heat to ensure complete solubility before cooling the solution.
- Maintain the reaction temperature between -5°C and 25°C and add an alkali metal carbonate catalyst (e.g., sodium carbonate, potassium carbonate) while stirring rapidly to initiate simultaneous deacetylation and Cbz removal.
- Upon completion, filter off solid byproducts, neutralize the filtrate with dilute acetic acid if necessary, and concentrate under reduced pressure followed by crystallization from an acetone-water mixture to isolate the pure intermediate.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain leaders, the adoption of this novel synthesis method translates into tangible strategic advantages that extend beyond simple yield metrics. The elimination of the hydrogenation step removes a major bottleneck in production scheduling, as hydrogenation reactors are often shared resources that require lengthy changeover times and safety certifications. By shifting to a simple stirred-tank reaction at ambient pressure, manufacturers can significantly increase asset utilization and reduce the lead time for producing high-purity pharmaceutical intermediates. Furthermore, the replacement of expensive palladium catalysts and anhydrous zinc salts with commodity alkali carbonates results in a drastic reduction in raw material costs. This substitution also mitigates supply chain risks associated with the volatility of precious metal prices and the geopolitical constraints on critical mineral sourcing.
- Cost Reduction in Manufacturing: The economic impact of this process is profound, primarily driven by the consolidation of two distinct reaction steps into one. This reduction in unit operations leads to substantial savings in labor, solvent consumption, and energy usage, as the energy-intensive reflux and high-pressure hydrogenation stages are completely obviated. Additionally, the avoidance of palladium on carbon eliminates the need for costly catalyst recovery systems or the financial loss associated with single-use noble metal catalysts. The simplified workup procedure, which relies on filtration and crystallization rather than complex extractions or column chromatography, further drives down the variable cost per kilogram, making the cost reduction in pharmaceutical intermediates manufacturing a reality rather than just a projection.
- Enhanced Supply Chain Reliability: From a logistics and planning perspective, the robustness of this chemistry ensures a more reliable supply of the critical intermediate. The reagents required, such as sodium carbonate and methanol, are globally available commodity chemicals with stable supply chains, unlike specialized catalysts that may face availability issues. The mild reaction conditions (-5°C to 25°C) are easily maintainable in standard glass-lined or stainless steel reactors without the need for specialized cryogenic or high-pressure infrastructure, allowing for greater flexibility in manufacturing site selection. This operational simplicity reduces the risk of batch failures due to equipment malfunction or operator error, thereby ensuring consistent delivery schedules for downstream API synthesis.
- Scalability and Environmental Compliance: Scaling this process from pilot plant to commercial production is inherently safer and more environmentally sustainable. The absence of hydrogen gas removes the explosion hazard, simplifying the safety case for large-scale operations and reducing insurance and compliance costs. Moreover, the process generates significantly less hazardous waste; the primary byproducts are benign alcohols and carbon dioxide, and the absence of heavy metals simplifies wastewater treatment and effluent discharge compliance. This alignment with green chemistry principles not only reduces environmental fees but also enhances the corporate sustainability profile of the manufacturing organization, which is increasingly important for partnerships with major multinational pharmaceutical companies.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this novel synthesis technology. These insights are derived directly from the experimental data and comparative examples provided in the patent literature, offering a transparent view of the process capabilities and limitations. Understanding these details is crucial for technical teams evaluating the feasibility of technology transfer and for commercial teams negotiating supply agreements.
Q: How does the new alkali carbonate method improve yield compared to traditional zinc acetate routes?
A: Traditional methods utilizing anhydrous zinc acetate followed by hydrogenation often suffer from cumulative yield losses, typically resulting in a total yield of less than 40%. The novel alkali carbonate method consolidates these steps into a single reaction, achieving isolated yields ranging from 72% to 89%, thereby significantly maximizing material throughput.
Q: What are the safety advantages of eliminating the hydrogenation step in Teniposide synthesis?
A: By removing the requirement for palladium-on-carbon catalyzed hydrogenation, the process eliminates the risks associated with high-pressure hydrogen gas and pyrophoric catalysts. This shift to ambient pressure alkali treatment drastically reduces operational hazards and simplifies the safety protocols required for industrial manufacturing.
Q: Does the mild alkaline condition affect the sensitive lactone or acetal structures in the molecule?
A: No, the process is designed to be highly chemoselective. By carefully controlling the temperature between -5°C and 25°C and using specific alkali carbonates, the reaction selectively removes acetyl and benzyloxycarbonyl groups without hydrolyzing the critical lactone ring or breaking the glycosidic bond, ensuring high structural integrity of the final product.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Teniposide Intermediate Supplier
At NINGBO INNO PHARMCHEM, we recognize that the efficient production of complex oncology intermediates requires not just chemical expertise but also a deep commitment to quality and scalability. As a leading CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that our clients receive a consistent and reliable supply of critical materials. Our state-of-the-art facilities are equipped to handle the mild alkaline conditions and crystallization processes described in this patent, adhering to stringent purity specifications and rigorous QC labs to guarantee that every batch meets the highest international standards for pharmaceutical use.
We invite potential partners to engage with our technical team to explore how this advanced synthesis route can optimize your supply chain for Teniposide and related derivatives. By leveraging our manufacturing capabilities, you can achieve a Customized Cost-Saving Analysis tailored to your specific volume requirements. We encourage you to contact our technical procurement team today to request specific COA data and route feasibility assessments, and let us demonstrate how our commitment to innovation can drive value for your organization.