Optimizing Kinase Inhibitor Synthesis: A Technical Breakthrough in Aniline Intermediate Manufacturing
Optimizing Kinase Inhibitor Synthesis: A Technical Breakthrough in Aniline Intermediate Manufacturing
The pharmaceutical industry continuously seeks robust synthetic pathways for complex heterocyclic intermediates, particularly those serving as key building blocks for tyrosine kinase inhibitors. Patent CN102174020B introduces a transformative methodology for the preparation of 5-(4-methyl-1H-imidazol-1-yl)-3-(trifluoromethyl)-aniline, a critical precursor in the synthesis of substituted pyrimidylaminobenzamides. This compound class has demonstrated significant potential in treating neoplastic diseases, including leukemia, by inhibiting kinases such as c-Abl and VEGF-R. The disclosed technology addresses long-standing challenges in yield consistency, safety, and scalability, offering a viable solution for reliable pharmaceutical intermediate supplier networks aiming to secure high-quality raw materials for oncology drug development pipelines.
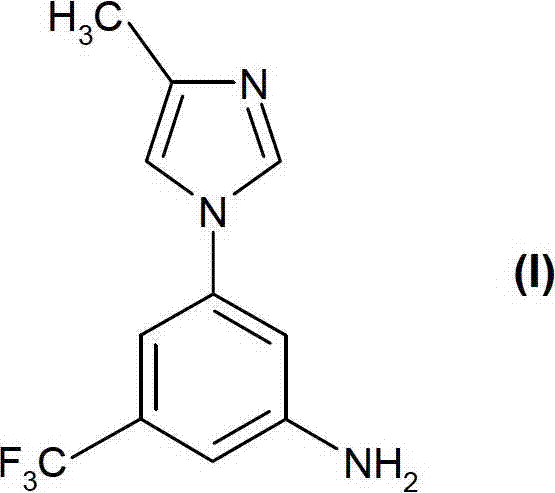
Historically, the synthesis of such fluorinated aniline derivatives relied on energetically demanding conditions that compromised both safety and economic efficiency. Conventional routes often necessitated high-temperature aromatic substitutions exceeding 150°C, leading to inconsistent product quality and significant formation of decomposition by-products. Furthermore, traditional methods frequently employed hazardous reagents like diphenylphosphoryl azide for Curtius rearrangements, creating substantial handling risks and requiring expensive chromatographic purification steps that are untenable for commercial operations. These limitations underscore the urgent need for cost reduction in anti-cancer drug manufacturing, driving the adoption of safer, more direct synthetic strategies that minimize waste and maximize throughput without sacrificing molecular integrity.
The Limitations of Conventional Methods vs. The Novel Approach
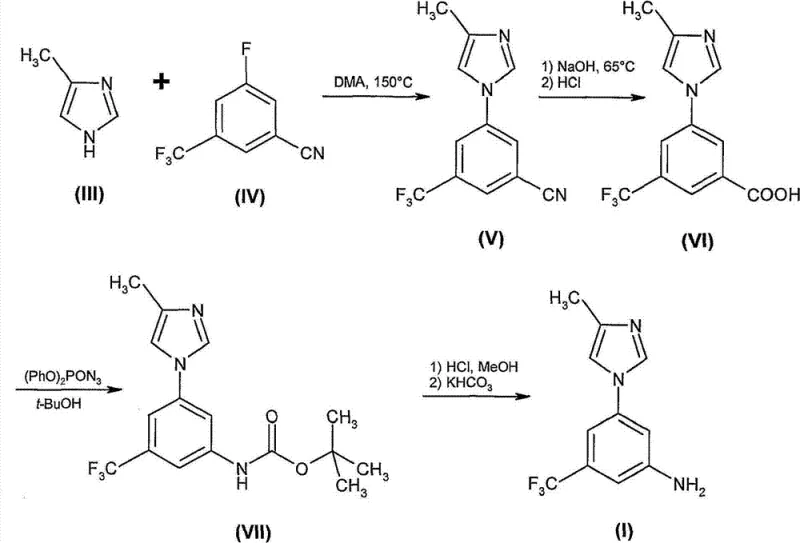
The Limitations of Conventional Methods
Prior art methodologies, such as the four-step route depicted in Scheme 1, initiated with an aromatic substitution reaction between 4-methylimidazole and 3-fluoro-5-trifluoromethylbenzonitrile under extreme thermal stress. This approach not only consumed excessive energy but also resulted in variable yields due to the harsh reaction environment which promoted side reactions. Subsequent conversion of the nitrile to the carboxylic acid and then to the amine via a Curtius rearrangement introduced significant complexity, requiring the use of toxic azide reagents that pose severe safety hazards in large-scale production. The final purification often demanded column chromatography, a technique that is prohibitively expensive and slow for industrial scale-up, thereby increasing lead time for high-purity aniline derivatives and creating bottlenecks in the supply chain for downstream API manufacturers.
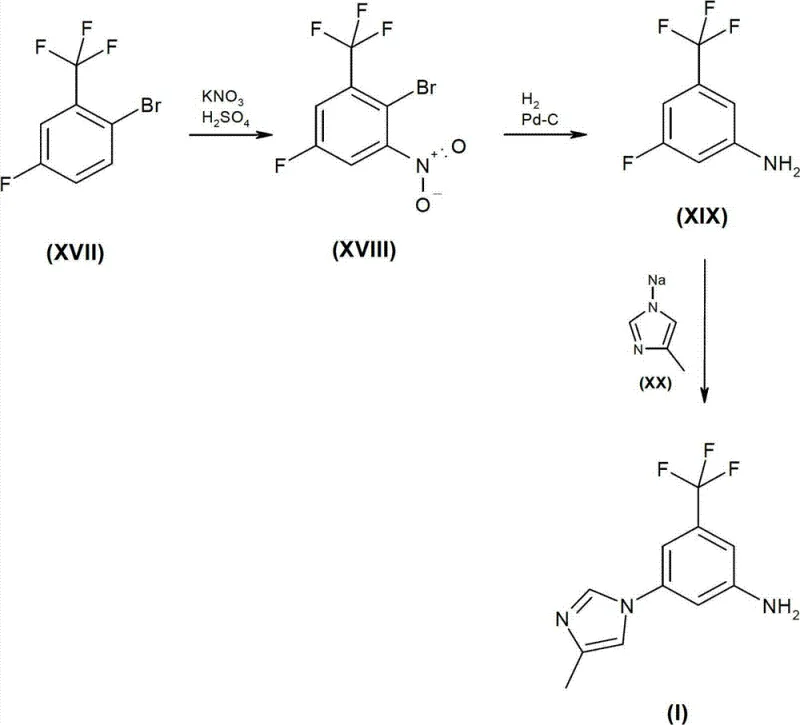
The Novel Approach
In stark contrast, the innovative process outlined in patent CN102174020B leverages a strategic sequence of nitration, reduction, and nucleophilic substitution to bypass these historical inefficiencies. By utilizing readily available starting materials such as 2-bromo-5-fluoro-alpha,alpha,alpha-trifluorotoluene, the new route enables the introduction of the nitro group under controlled acidic conditions, followed by selective reduction to the amine. This pathway avoids the use of high-energy substitution conditions and hazardous azide chemistry entirely. The resulting intermediate can be coupled with 4-methylimidazole using transition metal catalysts or direct nucleophilic displacement under milder basic conditions, significantly simplifying the workflow. This shift represents a paradigm change in commercial scale-up of complex heterocyclic compounds, offering a cleaner, safer, and more economically viable production model.
Mechanistic Insights into Transition Metal Catalyzed Coupling
The core of this technological advancement lies in the precise control of reaction mechanisms, particularly during the coupling of the imidazole moiety with the fluorinated benzene ring. When employing transition metal catalysts such as copper or palladium salts, the reaction proceeds through a coordinated oxidative addition and reductive elimination cycle that facilitates the formation of the C-N bond with high regioselectivity. The presence of ligands like Xantphos or cyclohexanediamine further stabilizes the catalytic species, ensuring that the reaction proceeds efficiently even at moderate temperatures ranging from 100°C to 150°C. This mechanistic precision minimizes the formation of constitutional isomers, a common impurity in such substitutions, thereby enhancing the overall purity profile of the final intermediate without the need for extensive downstream purification efforts.
Impurity control is further achieved through the strategic selection of reducing agents and solvents during the nitro-to-amine conversion step. Catalytic hydrogenation using palladium on carbon in alcoholic solvents allows for the selective reduction of the nitro group while preserving other sensitive functional groups like halogens. This selectivity is crucial for maintaining the structural integrity required for subsequent coupling reactions. Additionally, the process incorporates crystallization steps from solvents like toluene or heptane, which effectively exclude residual metals and organic by-products. Such rigorous control over the impurity profile ensures that the final product meets stringent purity specifications, a key requirement for R&D directors evaluating the feasibility of integrating this intermediate into GMP-compliant API synthesis workflows.
How to Synthesize 5-(4-Methyl-1H-Imidazol-1-yl)-3-Trifluoromethyl-Aniline Efficiently
The practical implementation of this synthesis involves a series of well-defined unit operations designed for reproducibility and safety. Initially, the nitration of the bromo-fluoro-toluene derivative is conducted in a dichloromethane medium with mixed acid, maintaining temperatures between 25°C and 40°C to prevent over-nitration. Following isolation, the nitro-intermediate undergoes catalytic hydrogenation, where careful monitoring of hydrogen uptake ensures complete conversion. The resulting amine is then reacted with 4-methylimidazole in a polar aprotic solvent like NMP or DMF, using a base such as sodium hydride or potassium carbonate to drive the substitution. Detailed standardized synthesis steps are provided in the guide below to assist technical teams in replicating these results.
- Preparation of nitro-intermediates via controlled nitration of bromo-fluoro-benzene derivatives using mixed acid systems.
- Catalytic hydrogenation or chemical reduction to convert nitro groups to amines while preserving sensitive halogen substituents.
- Final coupling with 4-methylimidazole using transition metal catalysts or nucleophilic aromatic substitution under basic conditions.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this novel synthetic route offers tangible benefits that extend beyond mere chemical elegance. By eliminating the need for hazardous azide reagents and high-energy reaction conditions, the process inherently reduces operational risks and associated insurance costs. The avoidance of chromatographic purification in favor of crystallization significantly lowers processing time and solvent consumption, leading to substantial cost savings in manufacturing overheads. Furthermore, the use of commercially available starting materials enhances supply chain reliability, reducing dependency on custom-synthesized precursors that often suffer from long lead times and availability issues.
- Cost Reduction in Manufacturing: The streamlined process eliminates expensive and hazardous reagents like diphenylphosphoryl azide, which require specialized handling and disposal protocols. By replacing chromatographic purification with efficient crystallization steps, the method drastically reduces solvent usage and processing time, leading to significant operational expenditure savings. The higher consistency in yield also minimizes raw material waste, optimizing the overall cost structure for producing high-purity kinase inhibitor intermediates.
- Enhanced Supply Chain Reliability: Utilizing widely available commodity chemicals such as 2-bromo-5-fluoro-alpha,alpha,alpha-trifluorotoluene ensures a stable supply of starting materials, mitigating the risk of production delays caused by precursor shortages. The robustness of the reaction conditions allows for flexible scheduling and faster turnaround times, enabling suppliers to respond more agilely to fluctuating market demands. This stability is critical for maintaining continuous API production schedules in the competitive oncology therapeutic sector.
- Scalability and Environmental Compliance: The process is designed with scalability in mind, avoiding unit operations that are difficult to translate from lab to plant, such as flash chromatography. The reduction in hazardous waste generation aligns with increasingly strict environmental regulations, simplifying compliance and reducing waste disposal costs. This green chemistry approach not only improves the corporate sustainability profile but also future-proofs the manufacturing process against tightening regulatory frameworks regarding chemical safety and emissions.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. They are derived from the specific pain points identified in prior art and the beneficial effects documented in the patent literature. Understanding these aspects is essential for stakeholders evaluating the integration of this intermediate into their existing supply chains.
Q: How does this new synthesis route improve upon conventional methods regarding safety?
A: The novel method eliminates the use of hazardous diphenylphosphoryl azide (DPPA) and high-energy aromatic substitution conditions (150°C), significantly reducing operational risks and decomposition by-products.
Q: What purity levels can be achieved with the described crystallization processes?
A: Through optimized recrystallization from toluene or heptane systems, the process consistently delivers intermediates with HPLC purity exceeding 99%, suitable for stringent GMP pharmaceutical applications.
Q: Is the process scalable for commercial production of kinase inhibitor precursors?
A: Yes, the route utilizes commercially available starting materials and avoids chromatographic purification steps, making it highly amenable to large-scale batch processing and continuous flow manufacturing.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 5-(4-Methyl-1H-Imidazol-1-yl)-3-Trifluoromethyl-Aniline Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that high-quality intermediates play in the successful development of life-saving medications. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the theoretical benefits of patent CN102174020B are fully realized in practical manufacturing environments. We adhere to stringent purity specifications and operate rigorous QC labs to guarantee that every batch of 5-(4-methyl-1H-imidazol-1-yl)-3-trifluoromethyl-aniline meets the exacting standards required by global pharmaceutical companies.
We invite you to collaborate with us to optimize your supply chain for kinase inhibitor production. Our technical procurement team is ready to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements. Contact us today to request specific COA data and route feasibility assessments, and let us demonstrate how our advanced manufacturing capabilities can support your drug development goals with reliability and precision.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →