Advanced Manufacturing of Fexofenadine Intermediates: A High-Yield Industrial Solution
Introduction to Advanced Intermediate Synthesis
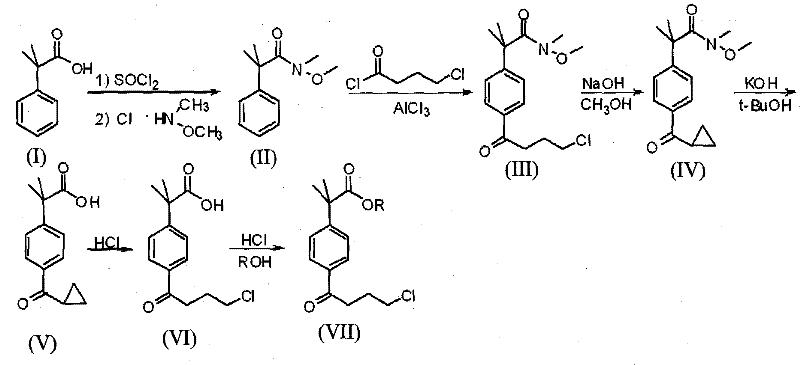
The pharmaceutical industry constantly seeks robust manufacturing pathways for critical antihistamine intermediates, particularly for second-generation drugs like fexofenadine hydrochloride. Patent CN101585767B introduces a transformative methodology for synthesizing 2-[4-(4-chlorobutyryl)phenyl]-2-methylpropionate, a pivotal building block in allergy medication production. This technical disclosure addresses long-standing inefficiencies in prior art by replacing hazardous, low-yield oxidation and reduction sequences with a streamlined cyclopropanation and ring-opening strategy. By shifting the synthetic logic away from direct Friedel-Crafts acylation of esters, which notoriously produces inseparable meta-isomers, this invention establishes a new benchmark for purity and operational safety. The protocol leverages readily available alkali metal hydroxides and alcohol solvents to achieve conversions that were previously unattainable without complex chromatographic purification. For global supply chain stakeholders, this represents a significant opportunity to secure a more reliable pharmaceutical intermediate supplier capable of delivering consistent quality without the volatility associated with older, pollution-heavy chemistries.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the production of 2-[4-(4-chlorobutyryl)phenyl]-2-methylpropionic acid esters has been plagued by severe structural selectivity issues inherent to classical electrophilic aromatic substitution. Traditional routes, such as those disclosed in U.S. Pat 6242606B1, rely on Friedel-Crafts acylation of 2-methyl-2-phenylpropionic acid esters, a process that inevitably generates substantial quantities of meta-isomer impurities alongside the desired para-product. These structural analogs possess nearly identical physical properties, rendering their separation via standard crystallization or distillation extremely difficult and cost-prohibitive, ultimately depressing the final product purity to unacceptable levels for GMP manufacturing. Furthermore, alternative pathways described in literature often necessitate the use of dangerous reagents such as lithium aluminum hydride for reduction steps and potassium permanganate for oxidation, introducing significant safety hazards and heavy metal contamination risks that complicate waste stream management. The cumulative effect of these inefficiencies is a fragmented supply chain where yields rarely exceed 40%, forcing manufacturers to process excessive volumes of raw materials to meet demand, thereby inflating costs and environmental footprints simultaneously.
The Novel Approach
In stark contrast, the methodology outlined in CN101585767B circumvents these regioselectivity pitfalls by employing a Weinreb amide intermediate that undergoes a controlled cyclopropanation followed by a specific acid-mediated ring opening. This strategic pivot allows for the construction of the chlorobutyryl side chain with absolute precision, ensuring that the final product is virtually free of the troublesome meta-isomers that plague conventional syntheses. The process initiates with the reaction of N-methyl-N-methoxyl-2-[4-(4-chlorobutyryl)phenyl]-2-methyl propanamide in an alkaline alcohol medium, forming a stable cyclopropyl ketone intermediate that serves as a protected precursor. Subsequent hydrolysis and acid treatment cleanly unveil the target chlorobutyryl functionality without the need for harsh oxidants or pyrophoric reducing agents. This not only simplifies the unit operations required for purification but also drastically reduces the generation of toxic byproducts, aligning modern manufacturing with stringent environmental compliance standards while maximizing atom economy.
Mechanistic Insights into Cyclopropanation and Ring-Opening Strategy
The core innovation of this synthesis lies in the mechanistic elegance of the intramolecular cyclization and subsequent nucleophilic ring opening, which acts as a temporary masking strategy for the reactive chlorobutyryl chain. In the initial phase, the presence of a strong base, such as sodium hydroxide or potassium hydroxide, in an alcoholic solvent facilitates the deprotonation of the gamma-carbon relative to the carbonyl group within the amide structure. This generates a carbanion species that attacks the terminal carbon bearing the chlorine atom, displacing the halide and closing the three-membered cyclopropane ring. This cyclization is thermodynamically driven and kinetically favorable under mild temperatures ranging from 20°C to 50°C, preventing side reactions that might occur at higher thermal energies. The resulting N-methyl-N-methoxyl-2-(4-cyclopropyl carbonyl phenyl)-2-methyl propanamide is a robust intermediate that can be isolated in high purity, effectively locking the molecular architecture into the correct para-substitution pattern before the final functional group manipulation occurs.
Following the formation of the cyclopropyl ketone, the process proceeds through a hydrolysis step to convert the Weinreb amide into the corresponding carboxylic acid, utilizing alkaline conditions followed by careful pH adjustment to isolate the acid form. The final transformation involves the acid-catalyzed ring opening of the cyclopropane moiety using concentrated mineral acid, typically hydrochloric acid, at elevated temperatures between 60°C and 100°C. Under these conditions, the strained cyclopropane ring is susceptible to nucleophilic attack by the chloride ion, which regioselectively opens the ring to regenerate the 4-chlorobutyryl side chain. This mechanism ensures that the chlorine atom is reintroduced at the precise terminal position required for the final API structure, bypassing the need for external chlorinating agents that could introduce impurities. The entire sequence demonstrates exceptional control over reaction pathways, minimizing the formation of oligomeric byproducts and ensuring that the impurity profile remains well within the specifications required for downstream pharmaceutical processing.
How to Synthesize 2-[4-(4-chlorobutyryl)phenyl]-2-methylpropionate Efficiently
Implementing this synthesis requires precise control over stoichiometry and reaction times to maximize the yield of each transformation step. The process begins with the preparation of an alkaline alcohol solution, where the concentration of the base is carefully maintained between 2 to 3.4 mol/L to ensure efficient deprotonation without causing excessive degradation of the sensitive amide backbone. The subsequent addition of the starting amide must be performed dropwise to manage the exotherm and maintain homogeneity, followed by a prolonged stirring period of 10 to 30 hours to drive the cyclopropanation to completion. Detailed standardized operating procedures for temperature ramping, solvent removal, and extraction protocols are critical for reproducibility. Adhering to these parameters allows manufacturers to consistently achieve yields exceeding 90% in the initial cyclization step, setting a strong foundation for the subsequent hydrolysis and ring-opening stages.
- Perform cyclopropanation of N-methyl-N-methoxyl-2-[4-(4-chlorobutyryl)phenyl]-2-methyl propanamide using alkali metal hydroxide in alcohol solvent at 20-50°C.
- Hydrolyze the resulting amide to 2-(4-cyclopropyl carbonyl phenyl)-2-methylpropanoic acid using alkaline alcohol solvent followed by pH adjustment.
- Execute ring-opening reaction with mineral acid at 60-100°C to obtain 2-[4-(4-chlorobutyryl)phenyl]-2-methylpropanoic acid.
- Finalize synthesis via esterification with hydrogen chloride gas in absolute alcohol to yield the target ester product.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this patented methodology offers profound advantages in terms of cost stability and logistical reliability. By eliminating the need for expensive and hazardous reagents like lithium aluminum hydride and potassium permanganate, the process significantly reduces the cost of goods sold associated with raw material procurement and specialized handling equipment. The removal of these dangerous chemicals also lowers insurance premiums and regulatory compliance costs, as the facility no longer needs to maintain extensive infrastructure for pyrophoric material storage or heavy metal waste disposal. Furthermore, the high selectivity of the reaction means that downstream purification steps are vastly simplified, reducing the consumption of solvents and energy required for recrystallization or chromatography, which translates directly into lower utility bills and faster batch cycle times.
- Cost Reduction in Manufacturing: The elimination of meta-isomer impurities removes the most costly bottleneck in traditional synthesis, which is the extensive purification required to meet pharmacopeial standards. Without the need to discard large fractions of off-spec material or run multiple recrystallizations to remove structural analogs, the effective yield of the process is substantially increased, leading to significant cost savings per kilogram of finished intermediate. Additionally, the use of commodity chemicals such as sodium hydroxide, methanol, and hydrochloric acid ensures that raw material costs remain low and predictable, shielding the supply chain from the price volatility often seen with specialty catalysts or organometallic reagents.
- Enhanced Supply Chain Reliability: The robustness of this chemical route ensures consistent batch-to-batch quality, which is critical for maintaining uninterrupted API production schedules. Because the process avoids complex multi-step sequences with low overall yields, the lead time for producing high-purity pharmaceutical intermediates is drastically shortened, allowing suppliers to respond more agilely to market demand fluctuations. The simplicity of the workup procedures, involving standard liquid-liquid extractions and filtrations, means that the process can be easily transferred between manufacturing sites without requiring highly specialized technical expertise, thereby diversifying the supplier base and mitigating single-source risks.
- Scalability and Environmental Compliance: From an environmental perspective, this method represents a greener alternative that aligns with increasingly strict global regulations on industrial emissions. The absence of heavy metal oxidants and the reduction in organic solvent usage due to higher purity profiles result in a much lighter environmental footprint, facilitating easier permitting and community relations for manufacturing plants. The process is inherently scalable, as the reaction conditions do not rely on delicate equilibrium states that are difficult to maintain in large reactors, making the commercial scale-up of complex pharmaceutical intermediates straightforward and economically viable for metric-ton production campaigns.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis route. Understanding these details is essential for R&D teams evaluating technology transfer and procurement officers assessing supplier capabilities. These insights are derived directly from the experimental data and claims within the patent documentation to ensure accuracy and relevance.
Q: How does this method improve upon traditional Friedel-Crafts acylation?
A: Unlike traditional Friedel-Crafts methods which generate difficult-to-separate meta-isomer impurities and suffer from low yields (around 27-40%), this novel route utilizes a cyclopropanation strategy that ensures high regioselectivity, effectively eliminating meta-isomers and significantly boosting overall yield to over 90% in key steps.
Q: What are the critical reaction conditions for the ring-opening step?
A: The ring-opening of the cyclopropane intermediate requires reaction with a mineral acid, specifically concentrated hydrochloric acid, at elevated temperatures ranging from 60°C to 100°C for 20 to 30 hours to ensure complete conversion to the chlorobutyryl derivative without degradation.
Q: Is this process suitable for large-scale commercial production?
A: Yes, the process is designed for industrial suitability by avoiding hazardous reagents like lithium aluminum hydride and potassium permanganate found in prior art, utilizing standard alkali metal hydroxides and alcohols, which simplifies waste treatment and enhances operational safety for metric-ton scale manufacturing.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 2-[4-(4-chlorobutyryl)phenyl]-2-methylpropionate Supplier
At NINGBO INNO PHARMCHEM, we recognize that the transition from laboratory discovery to commercial reality requires a partner with deep technical expertise and unwavering commitment to quality. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the sophisticated cyclopropanation chemistry described in CN101585767B can be executed with precision at an industrial level. We operate stringent purity specifications and maintain rigorous QC labs equipped with state-of-the-art analytical instrumentation to verify that every batch of 2-[4-(4-chlorobutyryl)phenyl]-2-methylpropionate meets the exacting standards required for fexofenadine synthesis, guaranteeing that your downstream processes remain unaffected by impurity variations.
We invite you to engage with our technical procurement team to discuss how this advanced manufacturing route can optimize your supply chain dynamics. By requesting a Customized Cost-Saving Analysis, you can gain a clear understanding of the economic benefits specific to your volume requirements. We encourage potential partners to contact us directly to索取 specific COA data and route feasibility assessments, allowing us to demonstrate our capability to deliver high-purity pharmaceutical intermediates with the reliability and transparency that global markets demand.