Advanced One-Pot Synthesis of 5'-Chloronucleosides for Commercial Pharmaceutical Manufacturing
The landscape of nucleoside chemistry is constantly evolving, driven by the urgent need for more efficient synthetic routes to critical antiviral and anticancer intermediates. Patent CN101250210B introduces a groundbreaking methodology for the preparation of 5'-chlorinated nucleosides, addressing long-standing inefficiencies in traditional halogenation protocols. This innovation utilizes a unique combination of triphenylphosphine and hexachloroethane to achieve selective chlorination at the 5'-position of nucleosides such as adenosine, guanosine, cytidine, and uridine. By leveraging a molar ratio of nucleoside to triphenylphosphine to hexachloroethane of 1:3-3.5:3-3.5, the process facilitates a seamless one-pot reaction that bypasses the cumbersome protection-deprotection sequences typically required in this chemical space. For R&D directors and procurement specialists seeking a reliable pharmaceutical intermediates supplier, this technology represents a significant leap forward in process intensification and cost efficiency.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of 5'-chloronucleosides has been plagued by operational complexity and environmental concerns that hinder scalable manufacturing. Traditional approaches often necessitate the rigorous protection of the 2' and 3' hydroxyl groups on the ribose sugar, as well as the amino groups on the nucleobase, to prevent unwanted side reactions and ensure regioselectivity. Literature references, such as those utilizing thionyl chloride (SOCl2) in pyridine, highlight severe pollution issues and the requirement for additional protective group manipulation when amino-bearing nucleosides are employed. Furthermore, alternative reagents like i-Pr2NCl combined with PPh3 have been reported, yet these methods suffer from prohibitively high reagent costs and lower overall atom economy. The cumulative effect of these multi-step protection and deprotection cycles is a drastic reduction in overall yield and a substantial increase in production time and waste generation, creating a bottleneck for the commercial scale-up of complex pharmaceutical intermediates.
The Novel Approach
In stark contrast to these legacy methods, the novel approach detailed in the patent employs a sophisticated in-situ protection strategy that streamlines the entire synthetic workflow into a single vessel. By reacting the starting nucleoside with triphenylphosphine and hexachloroethane, the system generates a reactive intermediate capable of selectively targeting the primary 5'-hydroxyl group while simultaneously shielding the secondary hydroxyls and exocyclic amines. This eliminates the need for external protecting groups, thereby simplifying the operational procedure to a mere dissolution, reflux, and workup sequence. The result is a robust process that operates under mild conditions, typically between 20°C and 50°C during the initial phase, followed by a moderate heating step at 70°C with acetic acid. This methodology not only enhances the purity profile of the final product but also drastically reduces the environmental footprint by avoiding hazardous chlorinating agents and excessive solvent usage associated with purification of protected intermediates.
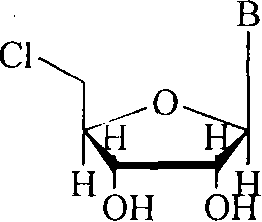
Mechanistic Insights into Triphenylphosphine-Hexachloroethane Mediated Chlorination
The core of this technological breakthrough lies in the unique mechanistic pathway facilitated by the interaction between triphenylphosphine (PPh3) and hexachloroethane (C2Cl6). Upon mixing, these reagents generate a transient phosphorus-chlorine species, effectively functioning as PPh3Cl6 in situ, which serves a dual purpose as both a chlorinating agent and a temporary protecting group donor. This intermediate reacts preferentially with the less sterically hindered primary 5'-hydroxyl group to form the chloro-substituted product, while concurrently forming phosphorus-nitrogen double bonds with amino groups on the nucleobase and stabilizing the 2' and 3' hydroxyls against chlorination. This intrinsic selectivity is the key to the method's success, as it prevents the formation of poly-chlorinated byproducts that are common in non-selective halogenation reactions. For technical teams evaluating route feasibility, understanding this mechanism is crucial, as it explains the high regioselectivity observed without the need for stoichiometric amounts of traditional protecting reagents.
Furthermore, the subsequent addition of acetic acid plays a pivotal role in the final liberation of the product and the quenching of phosphorus byproducts. The acidic environment facilitates the hydrolysis of any transient phosphate esters or phosphoramidates formed during the initial chlorination, ensuring that the final isolated material is the free 5'-chloronucleoside rather than a phosphorylated derivative. This step is critical for impurity control, as it ensures that the final API intermediate meets stringent purity specifications required for downstream coupling reactions in nucleotide prodrug synthesis. The ability to control the impurity profile through simple pH adjustment and temperature modulation demonstrates a level of process control that is highly desirable for GMP manufacturing environments, reducing the burden on downstream purification units.
How to Synthesize 5'-Chloronucleosides Efficiently
Implementing this synthesis route requires precise adherence to the reaction parameters outlined in the patent to maximize yield and minimize impurities. The process begins with the dissolution of the nucleoside substrate and triphenylphosphine in a suitable organic solvent such as dichloromethane, acetonitrile, or chloroform under an inert argon atmosphere to prevent oxidation of the phosphine reagent. Following this, hexachloroethane is added dropwise while maintaining reflux conditions, allowing the chlorinating species to form and react over a period of approximately 10 hours. The detailed standardized synthesis steps, including specific workup procedures and purification protocols via column chromatography, are provided in the guide below to assist your technical team in replicating these results.
- Dissolve the nucleoside substrate (adenosine, guanosine, cytidine, or uridine) and triphenylphosphine in an organic solvent such as dichloromethane or acetonitrile under argon protection.
- Add hexachloroethane dropwise under reflux conditions at temperatures between 20°C and 50°C, maintaining the reaction for approximately 10 hours to form the chlorinating intermediate.
- Cool the mixture to room temperature, add acetic acid, heat to 70°C for further reaction, and finally evaporate the solvent under reduced pressure to isolate the high-purity product.
Commercial Advantages for Procurement and Supply Chain Teams
From a strategic sourcing perspective, the adoption of this one-pot synthesis method offers profound advantages that directly impact the bottom line and supply chain resilience. By eliminating the discrete steps associated with protecting group chemistry, manufacturers can significantly reduce the consumption of raw materials, solvents, and energy, leading to substantial cost savings in pharmaceutical intermediates manufacturing. The simplification of the workflow also translates to a shorter overall cycle time, allowing for faster turnover of batches and improved responsiveness to market demand fluctuations. For supply chain heads focused on reducing lead time for high-purity pharmaceutical intermediates, this technology provides a more agile production model that mitigates the risks associated with complex multi-step syntheses.
- Cost Reduction in Manufacturing: The elimination of expensive protecting group reagents and the associated deprotection chemicals results in a direct reduction in the bill of materials. Furthermore, the high selectivity of the reaction minimizes the formation of difficult-to-remove impurities, which reduces the load on purification processes and increases the overall recovery of the final product. This efficiency gain allows for a more competitive pricing structure without compromising on quality, making it an attractive option for cost-sensitive projects.
- Enhanced Supply Chain Reliability: The use of readily available and stable reagents such as triphenylphosphine and hexachloroethane ensures a robust supply chain that is less susceptible to disruptions compared to methods relying on specialized or hazardous chlorinating agents. The mild reaction conditions also reduce the safety risks associated with exothermic events, facilitating smoother operations in standard chemical manufacturing facilities and ensuring consistent delivery schedules.
- Scalability and Environmental Compliance: The one-pot nature of the reaction significantly reduces the volume of waste generated per kilogram of product, aligning with modern green chemistry principles and regulatory requirements for waste disposal. The process is inherently scalable, as demonstrated by the patent's assertion of industrial value, allowing for seamless transition from pilot plant trials to full commercial production without the need for major equipment modifications or process re-engineering.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. These answers are derived directly from the experimental data and claims presented in the patent documentation, providing a factual basis for decision-making. Understanding these details is essential for evaluating the fit of this technology within your existing manufacturing portfolio.
Q: Does this synthesis method require protection of the 2' and 3' hydroxyl groups?
A: No, one of the primary advantages of this patented method is that the in-situ generated intermediate protects the 2' and 3' hydroxyl groups automatically, eliminating the need for separate protection and deprotection steps.
Q: What is the typical yield range for this 5'-chlorination process?
A: According to the patent data, the method consistently achieves high yields exceeding 83%, with specific examples demonstrating yields up to 95.8% depending on the specific nucleoside substrate used.
Q: Is this process suitable for large-scale industrial production?
A: Yes, the process is designed as a 'one-pot' synthesis with mild reaction conditions and simple workup procedures, making it highly scalable and industrially valuable for commercial manufacturing.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 5'-Chloronucleosides Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of efficient synthetic routes in the development of next-generation antiviral therapeutics. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from laboratory bench to industrial reactor is seamless and compliant. We are committed to delivering products that meet stringent purity specifications through our rigorous QC labs, guaranteeing that every batch of 5'-chloronucleosides performs consistently in your downstream synthesis applications.
We invite you to collaborate with us to leverage this advanced technology for your specific project needs. Please contact our technical procurement team to request a Customized Cost-Saving Analysis tailored to your volume requirements. We are ready to provide specific COA data and route feasibility assessments to demonstrate how our manufacturing capabilities can support your supply chain goals and accelerate your time to market.