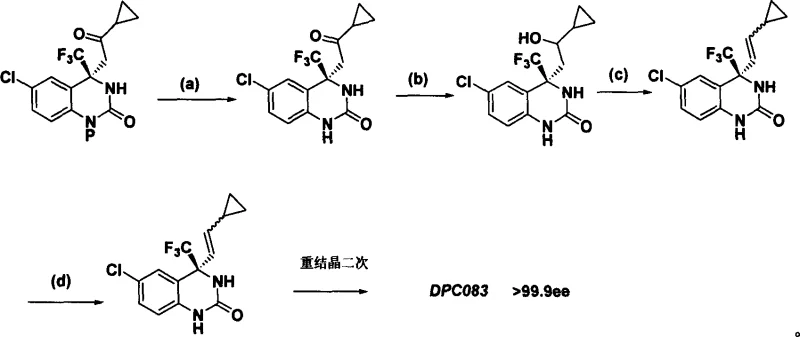
Scalable Organocatalytic Route for High-Purity Quinazolinone Pharmaceutical Intermediates
The pharmaceutical industry continuously seeks robust and scalable synthetic routes for complex active pharmaceutical ingredients (APIs) and their precursors. Patent CN1827605A introduces a groundbreaking methodology for the synthesis of 4,4-disubstituted-3,4-dihydro-2(1H)-quinazolinone compounds, which serve as critical intermediates in the production of non-nucleoside reverse transcriptase inhibitors (NNRTIs) like DPC083. This technology represents a significant paradigm shift from traditional metal-mediated processes to efficient organocatalytic systems. By leveraging small organic molecules to catalyze the asymmetric addition of methyl ketones to cyclic trifluoromethyl ketimine substrates, this invention addresses long-standing challenges in constructing chiral quaternary carbon centers. The ability to achieve high optical activity under mild reaction conditions offers substantial potential for industrial adoption, providing a safer and more cost-effective alternative to existing methodologies that rely on harsh cryogenic environments and stoichiometric amounts of hazardous reagents.
For procurement managers and supply chain directors, the implications of this technology extend beyond mere chemical novelty. The transition to organocatalysis eliminates the dependency on scarce and expensive transition metals, thereby mitigating supply chain risks associated with metal price volatility and availability. Furthermore, the simplified purification processes inherent to metal-free synthesis reduce the burden on waste management systems and lower the overall environmental footprint of the manufacturing process. This aligns perfectly with the growing global demand for green chemistry practices in pharmaceutical production. As a reliable pharmaceutical intermediate supplier, understanding these technological advancements is crucial for securing long-term supply contracts and ensuring the continuity of API production pipelines. The robustness of this synthetic route suggests a high degree of reproducibility, which is a key metric for quality assurance in regulated industries.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of DPC 961 and DPC 083 has relied on methods that present significant operational and economic hurdles. Traditional approaches often involve the fractional recrystallization of diastereomers or substrate-controlled diastereoselective 1,4-additions, which necessitate the use of chiral auxiliaries. These methods are inherently inefficient due to the theoretical maximum yield limit of 50% for resolution processes and the additional steps required to install and remove the auxiliary groups. Moreover, prior art such as WO 2001070707 describes chiral ligand-controlled asymmetric additions that require large excesses of both chiral ligands and strong bases like lithium alkyls and LHMDS. These reactions must be conducted at cryogenic temperatures, typically around minus 20°C, imposing severe energy costs and requiring specialized equipment capable of maintaining such low temperatures safely. The use of pyrophoric reagents also introduces significant safety hazards, complicating the scale-up process and increasing insurance and compliance costs for manufacturing facilities.
The Novel Approach
In stark contrast, the novel approach detailed in the patent utilizes an organic small molecule-catalyzed asymmetric addition reaction that operates under remarkably mild conditions. The reaction proceeds efficiently in aprotic organic solvents such as dimethyl sulfoxide (DMSO) at temperatures ranging from 0°C to 40°C, with room temperature being particularly recommended. This elimination of cryogenic requirements drastically simplifies the engineering controls needed for production. The catalytic system employs readily available amino acids like proline or specific diamine derivatives, which are far more economical and easier to handle than complex metal-ligand complexes. The process constructs the critical trifluoromethyl-containing quaternary carbon chiral center directly, avoiding the multi-step sequences associated with older methods. This streamlined pathway not only enhances the overall atom economy but also significantly reduces the generation of chemical waste, making it an environmentally superior choice for modern pharmaceutical manufacturing.
Mechanistic Insights into Organocatalytic Asymmetric Addition
The core of this technological advancement lies in the precise mechanistic interaction between the organic catalyst and the substrates. The reaction involves the nucleophilic attack of a methyl ketone, activated by the chiral amine catalyst, onto the electrophilic carbon of the cyclic trifluoromethyl ketimine. The catalyst, typically a proline derivative, forms a transient enamine or iminium intermediate with the ketone, thereby lowering the activation energy for the C-C bond formation while simultaneously imparting stereochemical control. This bifunctional activation ensures that the incoming nucleophile approaches the imine from a specific face, leading to the preferential formation of one enantiomer over the other. The presence of the trifluoromethyl group adds electronic complexity to the substrate, yet the catalytic system effectively manages this steric and electronic demand to produce the desired quaternary center with high fidelity. Understanding this mechanism is vital for R&D directors aiming to optimize reaction parameters such as solvent polarity and catalyst loading to maximize throughput.
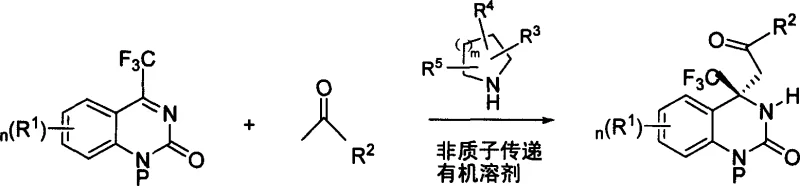
Impurity control is another critical aspect where this mechanism offers distinct advantages. In metal-catalyzed reactions, trace metal residues can persist in the final product, necessitating rigorous and costly purification steps to meet regulatory limits. Since this organocatalytic process is entirely metal-free, the impurity profile is significantly cleaner, primarily consisting of organic byproducts that are easier to separate via standard chromatographic or crystallization techniques. The patent highlights that initial enantiomeric excess values ranging from 70% to 80% ee can be achieved directly from the reaction, which can then be upgraded to greater than 99.9% ee through a single recrystallization step. This high level of stereochemical purity is essential for the biological activity of the final HIV inhibitor, as the wrong enantiomer could be inactive or even toxic. The robustness of the chiral induction mechanism ensures consistent quality across different batches, a key requirement for commercial API production.
How to Synthesize 4,4-Disubstituted-3,4-Dihydro-2(1H)-Quinazolinones Efficiently
The practical implementation of this synthesis involves dissolving the ketimine substrate and the methyl ketone in a suitable aprotic solvent, followed by the addition of the chiral catalyst. The reaction mixture is then stirred at ambient temperatures for a period ranging from 1 to 100 hours, depending on the specific substrates and catalyst loading. Monitoring can be performed using thin-layer chromatography (TLC) or by observing the color change of the reaction system from yellow to colorless. Upon completion, the product is isolated through aqueous workup and extraction with organic solvents like ethyl acetate, followed by purification via flash column chromatography or recrystallization. The detailed standardized synthesis steps see the guide below.
- Dissolve the cyclic trifluoromethyl ketimine substrate and the methyl ketone compound in an aprotic organic solvent such as DMSO.
- Add a metal-free organic catalyst, such as L-proline or D-proline, optionally with a dibasic acid co-catalyst.
- Stir the reaction mixture at mild temperatures ranging from 0°C to 40°C until completion, followed by standard workup and purification.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement specialists and supply chain leaders, the adoption of this organocatalytic technology translates into tangible strategic benefits that go beyond simple cost metrics. The primary advantage lies in the drastic simplification of the supply chain for raw materials. By replacing expensive and potentially hazardous metal catalysts and strong bases with commodity chemicals like proline and common ketones, manufacturers can source inputs from a broader and more stable vendor base. This diversification reduces the risk of supply disruptions caused by geopolitical issues or single-source dependencies. Additionally, the mild reaction conditions eliminate the need for specialized cryogenic reactors and the associated high energy consumption for cooling, leading to substantial operational expenditure savings. The reduced hazard profile also lowers insurance premiums and simplifies regulatory compliance regarding worker safety and environmental discharge.
- Cost Reduction in Manufacturing: The elimination of transition metals and stoichiometric chiral ligands removes a significant cost driver from the bill of materials. Traditional methods often require precious metals or complex ligands that are not only expensive to purchase but also difficult to recover and recycle. In this new process, the catalyst is used in catalytic amounts and is derived from abundant natural sources. Furthermore, the avoidance of cryogenic conditions means that standard stainless steel reactors can be used without the need for expensive cooling jackets or liquid nitrogen infrastructure. The simplified downstream processing, characterized by fewer purification steps and higher yields, further contributes to a lower cost of goods sold (COGS), allowing for more competitive pricing in the global market.
- Enhanced Supply Chain Reliability: Reliability is paramount in the pharmaceutical supply chain, where delays can halt clinical trials or commercial production. This synthesis route enhances reliability by utilizing robust and stable reagents that have long shelf lives and are not sensitive to minor fluctuations in temperature or moisture during storage. The scalability of the process is proven by its ability to tolerate a wide range of reaction times and concentrations without compromising product quality. This flexibility allows manufacturers to adjust production schedules dynamically in response to market demand without the fear of batch failures. The consistent quality of the intermediates produced ensures that downstream synthesis steps proceed smoothly, preventing bottlenecks that could delay the final API delivery.
- Scalability and Environmental Compliance: Scaling chemical processes from the laboratory to commercial production often reveals hidden challenges, but this organocatalytic method is inherently designed for scalability. The absence of exothermic hazards associated with strong bases and the use of benign solvents make the scale-up process predictable and safe. From an environmental perspective, the process aligns with green chemistry principles by minimizing waste generation and avoiding toxic heavy metals. This compliance with stringent environmental regulations facilitates faster approval from regulatory bodies and enhances the corporate social responsibility profile of the manufacturing entity. The ability to produce high-purity intermediates with a lower environmental footprint is increasingly becoming a deciding factor for partnerships with major pharmaceutical companies.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. These answers are derived directly from the experimental data and claims presented in the patent documentation, providing a factual basis for decision-making. Understanding these nuances helps stakeholders evaluate the feasibility of integrating this route into their existing manufacturing portfolios. The focus is on practical applicability, purity standards, and the specific advantages over legacy methods.
Q: What catalysts are suitable for this asymmetric addition reaction?
A: The patent specifies the use of organic small molecule catalysts, particularly proline derivatives like L-proline or D-proline, and specific diamine compounds, which avoid the need for expensive transition metals.
Q: How is high optical purity achieved in the final product?
A: High enantiomeric excess (ee) is initially achieved through the chiral catalyst, and further enhanced to >99.9% ee through a simple recrystallization process, ensuring pharmaceutical-grade purity.
Q: What are the advantages of this method over traditional metal-based synthesis?
A: This method operates under mild conditions (room temperature) without cryogenic cooling or strong bases like lithium alkyls, significantly improving safety and reducing operational costs.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 4,4-Disubstituted-3,4-Dihydro-2(1H)-Quinazolinone Supplier
At NINGBO INNO PHARMCHEM, we recognize the transformative potential of advanced organocatalytic technologies in the production of high-value pharmaceutical intermediates. Our team of expert chemists possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from laboratory discovery to industrial reality is seamless. We are committed to delivering products that meet stringent purity specifications, utilizing our rigorous QC labs to verify every batch against the highest international standards. Our infrastructure is designed to handle complex synthetic routes safely and efficiently, providing our partners with a secure and reliable source of critical building blocks for their drug development programs.
We invite you to collaborate with us to leverage this innovative synthesis route for your specific needs. Our technical procurement team is ready to provide a Customized Cost-Saving Analysis tailored to your project requirements, demonstrating how this technology can optimize your budget. Please contact us to request specific COA data and route feasibility assessments, and let us demonstrate how our expertise can accelerate your path to market while ensuring the highest quality and compliance.