Advanced Synthesis of Prasugrel Key Intermediate: A Cost-Effective and Scalable Route for API Manufacturing
Advanced Synthesis of Prasugrel Key Intermediate: A Cost-Effective and Scalable Route for API Manufacturing
The pharmaceutical industry continuously seeks robust and economically viable pathways for the production of critical active pharmaceutical ingredient (API) intermediates, particularly for high-volume drugs like Prasugrel, a potent antiplatelet agent. Patent CN102241612B introduces a significant technological advancement in the synthesis of 2-cyclopropyl-1-(2-fluorophenyl)-2-carbonyl ethyl p-toluenesulfonate, a pivotal building block in the Prasugrel value chain. This novel methodology departs from traditional, often problematic halogenation strategies by employing a sophisticated sequence involving Reimer-Tiemann carboxylation, Weinreb amide chemistry, and controlled Grignard addition. For R&D directors and procurement specialists, this patent represents a shift towards higher selectivity and reduced operational complexity, offering a compelling alternative to legacy processes that have long plagued supply chains with yield inconsistencies and purification challenges.
The strategic value of this synthesis lies in its foundational reliance on o-fluorobenzaldehyde, a readily accessible feedstock, rather than specialized ketone precursors that require complex upstream processing. By constructing the molecular architecture through a stepwise assembly—first establishing the acid framework, protecting the sensitive hydroxyl functionality, and then introducing the cyclopropyl group via nucleophilic attack—the process achieves a level of chemical precision that is difficult to replicate with direct functionalization methods. This approach not only enhances the overall purity profile of the final tosylate ester but also mitigates the formation of difficult-to-remove impurities that often arise from non-selective radical halogenation. Consequently, this technology stands out as a prime candidate for cost reduction in pharmaceutical intermediate manufacturing, aligning perfectly with the industry's drive towards leaner, more sustainable production models.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of alpha-halo ketones and their subsequent conversion to tosylates has been fraught with chemical inefficiencies. As illustrated in prior art such as Method One (WO2008108291A1), traditional routes often commence with the formation of a cyclopropyl ketone followed by a halogenation step at the alpha position. 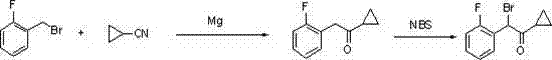 This halogenation step is notoriously difficult to control; the reactivity of the alpha-protons often leads to poly-halogenated byproducts, drastically reducing the yield of the desired mono-halogenated species. Furthermore, the use of aggressive halogenating agents necessitates stringent safety protocols and generates significant hazardous waste, complicating the environmental compliance profile of the manufacturing site. Other methods, such as Method Four, rely on oxidation reactions using isopropenyl acetate, which introduces expensive reagents and creates substantial environmental pollution, rendering them unsuitable for large-scale industrial application where cost and sustainability are paramount.
This halogenation step is notoriously difficult to control; the reactivity of the alpha-protons often leads to poly-halogenated byproducts, drastically reducing the yield of the desired mono-halogenated species. Furthermore, the use of aggressive halogenating agents necessitates stringent safety protocols and generates significant hazardous waste, complicating the environmental compliance profile of the manufacturing site. Other methods, such as Method Four, rely on oxidation reactions using isopropenyl acetate, which introduces expensive reagents and creates substantial environmental pollution, rendering them unsuitable for large-scale industrial application where cost and sustainability are paramount.
The Novel Approach
In stark contrast, the methodology disclosed in CN102241612B circumvents these pitfalls by reversing the logical order of bond formation. Instead of attempting to functionalize an existing ketone, the process builds the ketone functionality itself through a highly controlled Grignard reaction on a Weinreb amide intermediate. This strategy ensures that the carbonyl group is introduced with absolute regiochemical fidelity, eliminating the risk of over-halogenation entirely. The use of a methoxymethyl (MOM) protecting group during the Grignard addition safeguards the adjacent stereocenter and prevents unwanted side reactions, a level of protection that is absent in simpler, direct routes. By shifting the complexity from a difficult purification problem (separating halo-isomers) to a manageable synthetic sequence, this novel approach delivers a product with superior purity and consistency, directly addressing the quality demands of high-purity pharmaceutical intermediates required for FDA-approved drug production.
Mechanistic Insights into Weinreb Amide Mediated Grignard Addition
The core chemical innovation of this process resides in the utilization of N-methoxy-N-methylamide (Weinreb amide) chemistry to facilitate the introduction of the cyclopropyl group. The transformation begins with the conversion of the protected acetic acid derivative into its corresponding Weinreb amide using tris(dimethylamino)phosphine or similar amidation reagents. 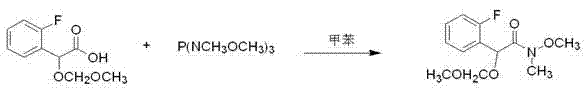 This specific amide functionality is crucial because it forms a stable tetrahedral intermediate upon reaction with the Grignard reagent (cyclopropylmagnesium bromide), preventing the over-addition that typically converts amides directly into tertiary alcohols. The chelation of the magnesium ion by both the carbonyl oxygen and the methoxy nitrogen locks the intermediate in a state that is stable until acidic workup, at which point it collapses cleanly to release the desired ketone. This mechanism provides a robust handle for C-C bond formation that is far more reliable than direct acylation or alkylation methods.
This specific amide functionality is crucial because it forms a stable tetrahedral intermediate upon reaction with the Grignard reagent (cyclopropylmagnesium bromide), preventing the over-addition that typically converts amides directly into tertiary alcohols. The chelation of the magnesium ion by both the carbonyl oxygen and the methoxy nitrogen locks the intermediate in a state that is stable until acidic workup, at which point it collapses cleanly to release the desired ketone. This mechanism provides a robust handle for C-C bond formation that is far more reliable than direct acylation or alkylation methods.
Furthermore, the stability of the methoxymethyl (MOM) ether protecting group throughout this rigorous organometallic sequence is a testament to the careful design of the synthetic route. During the Grignard reaction, performed in tetrahydrofuran (THF) at ambient temperatures, the MOM group remains inert, shielding the benzylic hydroxyl equivalent from nucleophilic attack or elimination. 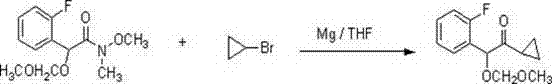 Only after the carbon skeleton is fully assembled is the protecting group removed under mild acidic conditions (HCl in methanol), revealing the alpha-hydroxy ketone motif essential for the final biological activity. This orthogonal protection strategy allows chemists to manipulate the oxidation state of the molecule without compromising the integrity of the sensitive fluorophenyl ring or the cyclopropyl moiety, ensuring a clean impurity profile that simplifies downstream processing.
Only after the carbon skeleton is fully assembled is the protecting group removed under mild acidic conditions (HCl in methanol), revealing the alpha-hydroxy ketone motif essential for the final biological activity. This orthogonal protection strategy allows chemists to manipulate the oxidation state of the molecule without compromising the integrity of the sensitive fluorophenyl ring or the cyclopropyl moiety, ensuring a clean impurity profile that simplifies downstream processing.
How to Synthesize 2-cyclopropyl-1-(2-fluorophenyl)-2-carbonyl Ethyl p-Toluenesulfonate Efficiently
The execution of this synthesis requires precise control over reaction parameters, particularly during the phase-transfer catalyzed carboxylation and the subsequent organometallic steps. The process initiates with the conversion of o-fluorobenzaldehyde to o-fluoromandelic acid using chloroform and aqueous sodium hydroxide, a reaction that demands efficient mixing and temperature control to maximize yield. Following esterification and MOM protection, the resulting acid is activated and converted into the Weinreb amide, setting the stage for the critical Grignard addition. The final stages involve the deprotection of the hydroxyl group and its conversion into the leaving group (tosylate) using p-toluenesulfonyl chloride in the presence of a base like triethylamine.  For detailed operational parameters, stoichiometry, and workup procedures, please refer to the standardized protocol below.
For detailed operational parameters, stoichiometry, and workup procedures, please refer to the standardized protocol below.
- Synthesize o-fluoromandelic acid from o-fluorobenzaldehyde using chloroform and sodium hydroxide under phase-transfer catalysis.
- Convert the acid to a Weinreb amide derivative via esterification, MOM protection, hydrolysis, and amidation with P(NMeOMe)3.
- Perform Grignard reaction with cyclopropylmagnesium bromide, followed by deprotection and tosylation to yield the final product.
Commercial Advantages for Procurement and Supply Chain Teams
From a commercial perspective, the adoption of this synthetic route offers transformative benefits for supply chain resilience and cost management. The primary advantage stems from the substitution of exotic or hazardous reagents with commodity chemicals that are available in bulk quantities globally. By anchoring the synthesis on o-fluorobenzaldehyde, manufacturers can leverage established supply chains for aromatic aldehydes, thereby insulating production from the volatility associated with specialized ketone precursors. This shift not only stabilizes raw material costs but also significantly reduces the lead time for sourcing, as suppliers of basic fluorinated aromatics are far more numerous than those of complex halogenated ketones. Consequently, this method supports a more agile commercial scale-up of complex pharmaceutical intermediates, allowing producers to respond rapidly to market demand fluctuations without being bottlenecked by scarce reagents.
- Cost Reduction in Manufacturing: The economic impact of this process is driven by the elimination of expensive oxidants and the reduction of waste disposal costs associated with toxic halogenation byproducts. Traditional methods often require costly purification steps to remove poly-halogenated impurities, which consumes solvents and silica gel while lowering overall throughput. In contrast, the high selectivity of the Weinreb amide route minimizes the formation of side products, leading to a streamlined isolation process that requires fewer chromatographic separations. This efficiency translates directly into lower operating expenditures (OPEX) per kilogram of product, providing a substantial margin improvement for contract manufacturing organizations (CMOs) and API producers alike.
- Enhanced Supply Chain Reliability: The robustness of the reaction conditions contributes to a more predictable manufacturing schedule. Unlike radical halogenation reactions which can be sensitive to trace impurities or light exposure, the ionic reactions employed in this patent (esterification, amidation, Grignard) are well-understood and easily controlled in standard stainless steel reactors. This reliability reduces the risk of batch failures and off-spec material, ensuring a consistent flow of high-quality intermediate to downstream customers. For supply chain heads, this means fewer disruptions and a more dependable inventory of critical materials, which is essential for maintaining continuous API production lines.
- Scalability and Environmental Compliance: The environmental footprint of this synthesis is markedly lower than that of prior art methods, facilitating easier regulatory approval and community acceptance. By avoiding the use of dichlorohydantoin and other chlorinating agents that generate corrosive and toxic waste streams, the process simplifies effluent treatment requirements. The solvents used, such as THF, methanol, and ethyl acetate, are standard in the industry and can be efficiently recovered and recycled. This alignment with green chemistry principles not only reduces environmental liability but also future-proofs the manufacturing asset against tightening global environmental regulations, ensuring long-term operational viability.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthetic technology. These insights are derived directly from the experimental data and comparative analysis provided in the patent documentation, offering clarity on the practical implications of adopting this route for industrial production.
Q: Why is the Weinreb amide strategy preferred over direct ketone halogenation for this intermediate?
A: Direct halogenation of ketones (as seen in prior art) often suffers from poor regioselectivity and lower yields due to poly-halogenation side reactions. The Weinreb amide route allows for precise construction of the carbon skeleton via Grignard addition, ensuring the carbonyl group is formed exactly where needed without competing side reactions.
Q: What are the primary cost drivers in this synthesis compared to other methods?
A: This method utilizes o-fluorobenzaldehyde, a widely available and inexpensive commodity chemical, as the starting material. Unlike methods requiring expensive oxidants like isopropenyl acetate or toxic halogenating agents like dichlorohydantoin, this route relies on standard reagents (chloroform, NaOH, TsCl), significantly reducing raw material costs and waste disposal expenses.
Q: How does this process address environmental and safety concerns in large-scale manufacturing?
A: The process avoids the use of highly toxic halogenating reagents and heavy metal catalysts found in alternative routes. By employing milder conditions and generating less hazardous byproducts, the method simplifies wastewater treatment and reduces the burden on labor protection systems, making it more suitable for green chemistry compliance in modern facilities.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 2-cyclopropyl-1-(2-fluorophenyl)-2-carbonyl Ethyl p-Toluenesulfonate Supplier
At NINGBO INNO PHARMCHEM, we recognize that the transition from laboratory patent to commercial reality requires more than just chemical knowledge; it demands engineering excellence and rigorous quality assurance. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the intricate steps of this synthesis—from the moisture-sensitive Grignard reaction to the final crystallization—are executed with precision. We maintain stringent purity specifications and operate rigorous QC labs equipped with advanced analytical instrumentation to verify that every batch of 2-cyclopropyl-1-(2-fluorophenyl)-2-carbonyl ethyl p-toluenesulfonate meets the exacting standards required for global pharmaceutical markets.
We invite procurement leaders and technical directors to engage with us for a Customized Cost-Saving Analysis tailored to your specific volume requirements. By leveraging our optimized version of this patent technology, we can help you achieve significant reductions in COGS while securing a stable supply of this critical intermediate. Please contact our technical procurement team today to request specific COA data and route feasibility assessments, and let us demonstrate how our commitment to innovation can drive value for your organization.