Industrial Scale Synthesis of Beta-Halo-Amino Acids and S-Phenylcysteine Derivatives for HIV Inhibitors
Introduction to Advanced Amino Acid Halogenation Technology
The pharmaceutical industry constantly seeks more efficient pathways to synthesize complex chiral building blocks, particularly for antiretroviral therapies. Patent CN1283178A introduces a groundbreaking methodology for the preparation of beta-halo-alpha-aminocarboxylic acids and their derivatives, specifically targeting the synthesis of optically active N-protected-S-phenylcysteine. This technology addresses long-standing challenges in amino acid chemistry by enabling the direct halogenation of beta-hydroxy-alpha-aminocarboxylic acids, such as serine, without the need for prior carboxyl group protection. This represents a significant paradigm shift from traditional multi-step sequences, offering a robust solution for producing high-purity intermediates essential for HIV protease inhibitors. The ability to maintain high optical purity while simplifying the synthetic route provides a compelling value proposition for manufacturers aiming to optimize their supply chains for critical antiviral medications.
For R&D directors and process chemists, the implications of this patent extend beyond mere academic interest; it offers a tangible route to reduce process mass intensity and improve overall yield. The core innovation lies in treating the beta-hydroxy group with a halogenating agent in the presence of an acid, effectively bypassing the cumbersome esterification and hydrolysis steps that have historically plagued this chemical transformation. By leveraging readily available starting materials like L-serine or D-serine, this method ensures a consistent supply of chirally pure intermediates. As a reliable pharmaceutical intermediate supplier, understanding these mechanistic nuances is crucial for scaling production while adhering to stringent regulatory standards for impurity profiles and enantiomeric excess.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of beta-halo-alpha-aminocarboxylic acids has been fraught with inefficiencies that hinder industrial scalability. Conventional methodologies typically necessitate a three-step sequence: first, the derivatization of the beta-hydroxy-alpha-aminocarboxylic acid into a corresponding ester to protect the carboxyl functionality; second, the halogenation of the beta-hydroxyl group using phosphorus halides; and finally, the hydrolysis of the ester group to regenerate the free acid. This multi-step approach not only increases the operational complexity but also invariably leads to lower overall yields due to material losses at each isolation stage. Furthermore, the use of aggressive reagents and the requirement for strict anhydrous conditions during esterification add significant cost and safety burdens to the manufacturing process.
Alternative methods involving the use of thionyl chloride often require it to act as both reagent and solvent in large excess, complicating downstream processing and waste disposal. Additionally, previous attempts to synthesize S-phenylcysteine derivatives from serine often relied on enzymatic processes or complex lactonization strategies that suffered from poor operability and limited production capacity. The reliance on difficult-to-handle reagents like sodium hydride for substitution reactions further exacerbated safety concerns and often resulted in decreased optical purity due to racemization under harsh basic conditions. These cumulative drawbacks highlight the urgent need for a more streamlined, industrially advantageous process that can deliver high-quality intermediates without compromising on safety or efficiency.
The Novel Approach
The methodology disclosed in the patent revolutionizes this landscape by enabling the direct conversion of beta-hydroxy-alpha-aminocarboxylic acids into their beta-halo counterparts in a single reaction step. By treating the substrate with a halogenating agent, preferably thionyl chloride, in the presence of a hydrogen halide like hydrogen chloride gas, the process effectively activates the hydroxyl group for substitution without masking the amino group's basicity or protecting the carboxyl group. This direct approach drastically simplifies the workflow, eliminating the need for esterification and subsequent hydrolysis. The reaction can be conducted in common ether solvents such as 1,4-dioxane or tetrahydrofuran, facilitating easy solvent recovery and recycling, which is a key factor in reducing the environmental footprint of the synthesis.
Moreover, this novel route demonstrates exceptional compatibility with optically active starting materials, preserving the stereochemical integrity of the alpha-carbon throughout the transformation. The resulting beta-halo intermediates can be seamlessly converted into N-protected-S-phenylcysteine derivatives through subsequent protection and thiophenylation steps. This continuity allows for a telescoped process where intermediates need not be isolated in high purity before the next step, further enhancing throughput. For procurement managers, this translates to a more resilient supply chain capable of meeting the demanding volume requirements of the global antiviral market while maintaining competitive pricing structures through reduced operational overheads.
Mechanistic Insights into Direct Halogenation and Thiophenylation
The chemical elegance of this process lies in its ability to navigate the reactivity of multifunctional amino acids without extensive protecting group manipulation. The mechanism involves the activation of the beta-hydroxyl group by the halogenating agent, likely forming a chlorosulfite intermediate which is then displaced by a chloride ion. The presence of hydrogen chloride gas plays a dual role: it protonates the amino group to prevent unwanted side reactions while simultaneously driving the equilibrium towards the formation of the beta-chloro product. This careful balance ensures that the nucleophilic attack occurs selectively at the beta-position, leaving the alpha-stereocenter undisturbed. The use of specific solvents like 1,4-dioxane helps stabilize the transition state and solubilize the zwitterionic nature of the amino acid salts, ensuring homogeneous reaction conditions that are vital for consistent batch-to-batch reproducibility.
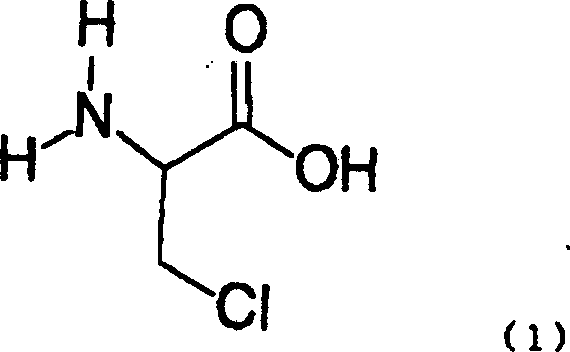
Following the halogenation, the resulting beta-chloroalanine serves as a versatile electrophile for the introduction of the phenylthio group. The subsequent thiophenylation step is conducted under controlled alkaline conditions using thiophenol. A critical aspect of this mechanism is the timing of the amino protection; the patent reveals that protecting the amino group prior to thiophenylation is essential to prevent decomposition of the beta-chloro intermediate under the basic conditions required for sulfur substitution. By employing protecting groups like benzyloxycarbonyl (Cbz), the amino functionality is masked, allowing the nucleophilic aromatic substitution to proceed efficiently. This strategic sequencing prevents the formation of aziridine byproducts and ensures that the final S-phenylcysteine derivative retains the high optical purity of the starting serine, a parameter that is non-negotiable for pharmaceutical applications.
Impurity control is inherently built into this mechanistic design. By avoiding the high temperatures and strong bases associated with older methods, the formation of elimination byproducts (such as dehydroalanine derivatives) is minimized. The isolation of the beta-halo acid as a hydrohalide salt allows for purification via crystallization from water-miscible organic solvents like acetone, effectively removing inorganic salts and unreacted starting materials. This solid-state purification strategy is far more scalable than column chromatography, which is often required for more complex, less stable intermediates. For quality assurance teams, this means a cleaner crude profile entering the final steps, reducing the burden on downstream purification and ensuring that the final API intermediate meets the rigorous specifications required for clinical use.
How to Synthesize Beta-Chloroalanine Efficiently
The practical implementation of this synthesis route involves a carefully orchestrated sequence of reactions designed to maximize yield and optical purity. The process begins with the suspension of optically active serine in a suitable ether solvent, followed by saturation with hydrogen chloride gas to form the acid salt. Thionyl chloride is then added, and the mixture is heated to facilitate the halogenation. Once the beta-chloroalanine is formed, it can be isolated or carried forward directly. The subsequent protection step involves adjusting the pH to alkaline conditions and adding an amino-protecting agent like benzyl chloroformate. Finally, the protected intermediate reacts with thiophenol to yield the target S-phenylcysteine derivative. Detailed standard operating procedures for each stage, including precise temperature controls and stoichiometric ratios, are essential for successful technology transfer.
- Treat optically active serine or its salt with a halogenating agent like thionyl chloride in the presence of hydrogen chloride gas to form beta-chloroalanine.
- React the resulting beta-chloroalanine with an amino-protecting agent such as benzyl chloroformate under basic conditions to form N-protected-beta-chloroalanine.
- Subject the N-protected intermediate to thiophenylation using thiophenol under alkaline conditions to yield the final optically active N-protected-S-phenylcysteine.
Commercial Advantages for Procurement and Supply Chain Teams
From a commercial perspective, the adoption of this patented technology offers substantial benefits for organizations managing the procurement of complex amino acid intermediates. The primary advantage lies in the drastic simplification of the manufacturing process, which directly correlates to reduced production costs. By eliminating the esterification and hydrolysis steps, manufacturers save on reagents, solvents, and energy consumption associated with heating and cooling cycles. This streamlining also reduces the total reactor occupancy time, allowing facilities to increase their production capacity without additional capital investment in hardware. For procurement managers negotiating contracts, this efficiency provides a buffer against raw material price volatility, ensuring more stable pricing for long-term supply agreements.
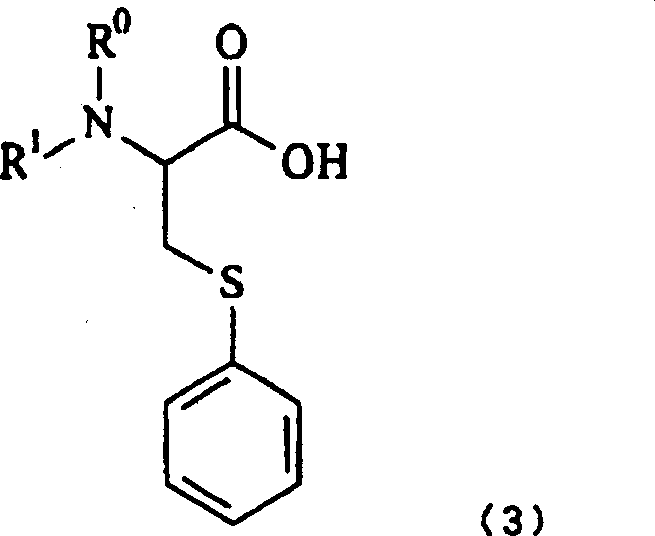
Supply chain reliability is another critical benefit derived from this methodology. The reliance on commodity chemicals like serine, thionyl chloride, and thiophenol means that the raw material base is broad and secure, minimizing the risk of supply disruptions caused by niche reagent shortages. Furthermore, the robustness of the reaction conditions allows for flexible manufacturing across different geographic locations, supporting a diversified supply chain strategy. The ability to produce high-purity intermediates with minimal racemization reduces the risk of batch rejection, ensuring a continuous flow of material to downstream API synthesis sites. This reliability is paramount for pharmaceutical companies managing just-in-time inventory systems for life-saving medications.
Environmental compliance and scalability are also significantly enhanced by this approach. The reduction in step count inherently lowers the volume of chemical waste generated, simplifying effluent treatment and reducing disposal costs. The use of recoverable solvents like 1,4-dioxane aligns with green chemistry principles, appealing to stakeholders focused on sustainability metrics. Scalability is proven by the patent's examples, which demonstrate successful execution from gram to multi-gram scales with consistent results. This suggests that the process can be readily adapted for commercial scale-up of complex pharmaceutical intermediates, meeting the tonnage requirements of major drug manufacturers while adhering to strict environmental regulations.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the synthesis of beta-halo-amino acids and S-phenylcysteine derivatives. These insights are derived directly from the technical specifications and experimental data provided in the patent literature, offering clarity on process feasibility and product quality. Understanding these details helps stakeholders make informed decisions about integrating this technology into their existing manufacturing portfolios.
Q: How does this new method improve optical purity compared to conventional routes?
A: The patented process avoids harsh conditions and multiple protection-deprotection cycles that typically cause racemization. By directly halogenating the beta-hydroxy group of serine without prior esterification, the method maintains the chiral integrity of the alpha-carbon, achieving optical purities exceeding 98% e.e., which is critical for the efficacy of HIV protease inhibitors.
Q: What are the primary cost drivers eliminated in this synthesis route?
A: Traditional methods require esterification of the carboxyl group followed by hydrolysis, adding two distinct reaction steps and associated solvent recovery costs. This novel approach eliminates the need for carboxyl protection and subsequent deprotection, significantly reducing raw material consumption, reactor occupancy time, and waste generation, thereby lowering the overall cost of goods sold for these complex intermediates.
Q: Is this process suitable for large-scale commercial manufacturing?
A: Yes, the patent explicitly describes conditions optimized for industrial advantage, utilizing common solvents like 1,4-dioxane and reagents like thionyl chloride. The isolation procedures involve simple crystallization and filtration rather than complex chromatography, making the technology highly transferable to multi-ton production scales required for global pharmaceutical supply chains.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Beta-Chloroalanine Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of high-quality intermediates in the development of next-generation therapeutics. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the innovative chemistry described in patent CN1283178A can be effectively translated into industrial reality. We are committed to delivering products with stringent purity specifications, utilizing our rigorous QC labs to verify optical purity and impurity profiles at every stage of production. Our capability to handle complex chiral synthesis positions us as a strategic partner for pharmaceutical companies seeking to secure their supply of HIV inhibitor intermediates.
We invite you to collaborate with us to explore how this advanced synthesis route can optimize your cost structure and enhance your supply chain resilience. Our technical procurement team is ready to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements. Please contact us to request specific COA data and route feasibility assessments, and let us demonstrate how our expertise in amino acid chemistry can drive value for your organization.