Mastering Etoricoxib Quality Control: Novel Synthesis of Critical Impurity Standards
Mastering Etoricoxib Quality Control: Novel Synthesis of Critical Impurity Standards
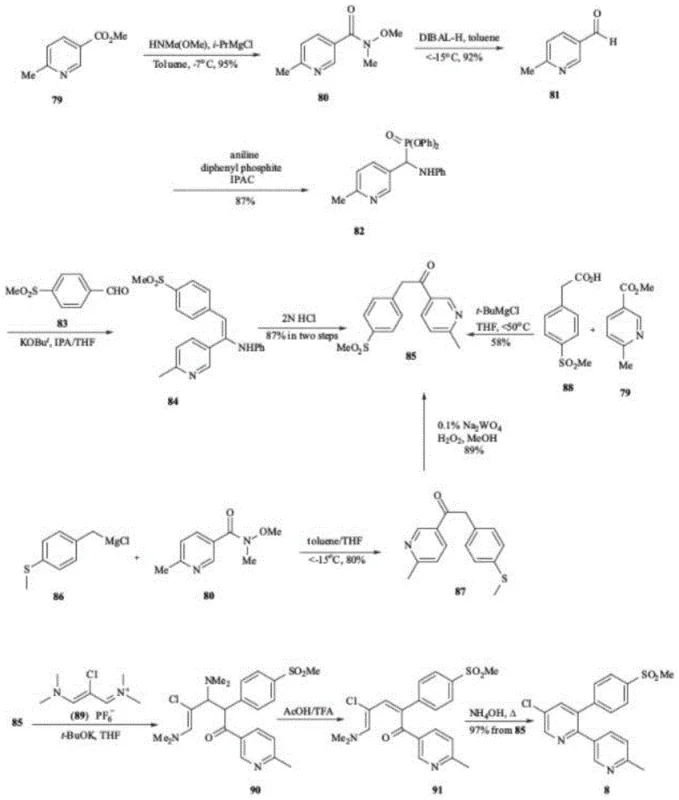
In the highly regulated landscape of pharmaceutical manufacturing, the integrity of Active Pharmaceutical Ingredients (APIs) is paramount. A recent technological breakthrough detailed in patent CN114989071A addresses a critical gap in the quality control framework for Etoricoxib, a widely prescribed selective COX-2 inhibitor. This patent introduces a robust preparation method for a previously unreported impurity, designated as Compound I, which arises during the synthesis of key intermediates. For R&D directors and quality assurance teams, the ability to synthesize and characterize this specific impurity with high precision is not merely an academic exercise; it is a strategic necessity for ensuring batch consistency and regulatory compliance. The disclosed method achieves an exceptional purity of 99.6%, meeting the rigorous standards required for reference substances in national formularies.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of Etoricoxib has relied on convergent routes involving the coupling of pyridine intermediates. While established protocols, such as those described in earlier patents like EP2551265, effectively produce the target API, they often leave behind a complex matrix of related substances. A significant limitation in previous quality control methodologies was the presence of "unknown" peaks in chromatographic profiles. Specifically, during the Grignard reaction steps used to construct the carbon skeleton, side reactions involving tert-butyl groups could generate structurally similar byproducts. Without a certified reference standard for these specific byproducts, manufacturers were forced to classify them as "unidentified impurities." This classification poses a severe regulatory risk, as agencies demand full characterization of impurities exceeding identification thresholds. The inability to distinguish Compound I from other degradation products or starting materials often led to conservative batch rejections or costly delays in stability testing programs.
The Novel Approach
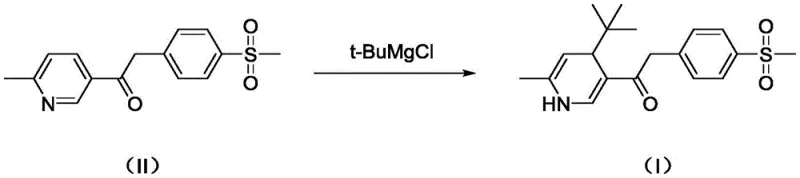
The methodology outlined in patent CN114989071A represents a paradigm shift from reactive quality control to proactive impurity management. Instead of attempting to suppress the formation of Compound I entirely—which may be thermodynamically favorable under certain process conditions—the inventors have developed a dedicated synthetic route to produce it intentionally. By isolating Compound I as a discrete, high-purity entity, pharmaceutical manufacturers can now spike their analytical samples with known quantities of this impurity. This allows for the precise calibration of HPLC detectors and the validation of separation methods. The novel approach utilizes a controlled Grignard addition of tert-butyl magnesium chloride to a specific ketone intermediate (Compound II). This targeted synthesis ensures that the reference material is chemically identical to the process impurity, thereby eliminating ambiguity in quality testing and providing a solid foundation for setting scientifically justified acceptance criteria.
Mechanistic Insights into Grignard-Mediated Impurity Formation
To fully appreciate the utility of this reference standard, one must understand the mechanistic pathway leading to Compound I. The formation of this impurity is intrinsically linked to the nucleophilic addition steps employed in the construction of the Etoricoxib backbone. In the standard synthesis of the key intermediate (Compound II), a ketone functionality is present which is susceptible to nucleophilic attack. When tert-butyl magnesium chloride (t-BuMgCl) is introduced—often intended for a different transformation or present as a reagent in parallel steps—it can act as a nucleophile attacking the carbonyl carbon of the pyridine-containing ketone. This addition results in the formation of a tertiary alcohol structure, which corresponds to the core scaffold of Impurity I. The steric bulk of the tert-butyl group significantly influences the reaction kinetics and the subsequent purification challenges. Understanding this mechanism allows process chemists to map the "design space" of the reaction, identifying exactly where in the process flow this impurity is most likely to generate.
Furthermore, the structural confirmation provided in the patent, including single-crystal X-ray diffraction data, offers definitive proof of the stereochemistry and connectivity of Compound I. This level of characterization is vital for distinguishing it from isomeric impurities that might exhibit similar retention times in chromatography but possess different toxicological profiles. The patent details the use of specific solvents like tetrahydrofuran (THF) and precise temperature controls (60°C) to drive this side reaction to completion for the purpose of standard generation. By mastering the kinetics of this Grignard addition, the inventors have turned a potential liability (an uncontrolled process impurity) into a valuable asset (a qualified reference standard). This mechanistic clarity empowers R&D teams to refine their main synthesis routes, potentially adjusting stoichiometry or addition rates to minimize the in-process formation of Compound I while simultaneously having the tools to detect it if it does occur.
How to Synthesize Etoricoxib Impurity I Efficiently
The preparation of this critical reference standard requires strict adherence to the optimized conditions described in the patent to ensure the highest possible purity. The process begins with the dissolution of the precursor ketone (Compound II) in an anhydrous etheral solvent, typically tetrahydrofuran, under an inert atmosphere to prevent moisture interference. The reaction temperature is carefully ramped to 60°C before the slow, dropwise addition of the Grignard reagent, tert-butyl magnesium chloride. This controlled addition is crucial to manage the exotherm and favor the formation of the desired tertiary alcohol over other potential side products. Following the reaction period, the mixture is quenched with dilute acetic acid, which protonates the alkoxide intermediate and precipitates the crude product. The final purification step involves silica gel column chromatography using a gradient elution system, which effectively separates the target impurity from unreacted starting materials and other closely related byproducts, yielding a white solid suitable for analytical use.
- Dissolve Compound II in tetrahydrofuran (THF) and heat the mixture to 60°C under inert atmosphere.
- Slowly add tert-butyl magnesium chloride (t-BuMgCl) solution dropwise while maintaining the temperature at 60°C for 1 hour.
- Quench the reaction with dilute acetic acid, isolate the precipitate, and purify via silica gel column chromatography to achieve >99% purity.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain leaders, the availability of high-quality impurity standards like Compound I translates directly into risk mitigation and operational efficiency. The primary value proposition lies in the acceleration of regulatory filings and the stabilization of the supply chain for the finished API. When a manufacturer can demonstrate to regulatory bodies that they have full control over their impurity profile—backed by authenticated reference standards—they significantly reduce the likelihood of audit observations or clinical hold placements. This certainty allows for more predictable production schedules and reduces the inventory buffers often held to account for potential batch failures. Moreover, sourcing these complex reference materials from a specialized supplier eliminates the need for internal R&D teams to divert resources toward synthesizing small quantities of impurities, allowing them to focus on process optimization and scale-up activities.
- Cost Reduction in Manufacturing: The implementation of validated analytical methods using certified reference standards leads to substantial cost savings by preventing false-positive results in quality control testing. Without a specific standard for Compound I, QC labs might flag batches as "out of specification" due to unidentified peaks, leading to expensive investigations, re-processing, or total batch scrapping. By accurately quantifying this impurity, manufacturers can prove that levels remain well within safe, approved limits, thereby salvaging valuable production runs. Additionally, the elimination of the need for internal, small-scale synthesis of these standards reduces the overhead associated with specialized hazardous reagents and dedicated purification equipment.
- Enhanced Supply Chain Reliability: Access to a reliable source of Etoricoxib impurity standards ensures continuity in long-term stability studies, which are mandatory for maintaining market authorization. Disruptions in the supply of reference materials can halt stability testing, delaying product renewals or variations. A stable supply of these critical reagents supports a resilient quality control infrastructure, ensuring that every batch of API released to the market meets the stringent specifications required by global health authorities. This reliability fosters stronger partnerships between API manufacturers and their downstream formulation partners, who depend on consistent quality documentation.
- Scalability and Environmental Compliance: The synthesis method described in the patent utilizes common laboratory reagents and standard purification techniques, making it amenable to scale-up should larger quantities of the reference standard be required for multi-site validation. Furthermore, by defining the impurity profile so precisely, waste streams from the main API production can be better characterized and managed. Knowing exactly what byproducts are present allows for more targeted waste treatment protocols, aligning with modern green chemistry initiatives and reducing the environmental footprint of pharmaceutical manufacturing operations.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the application and sourcing of Etoricoxib impurity standards. These insights are derived from the technical specifications and experimental data provided in the underlying patent literature, aiming to clarify the role of Compound I in the broader context of drug development and quality assurance.
Q: Why is the synthesis of Etoricoxib Impurity I critical for API manufacturing?
A: Impurity I is a newly identified byproduct formed during the Grignard step of Etoricoxib synthesis. Having a certified reference standard allows manufacturers to accurately quantify this impurity using HPLC, ensuring compliance with strict pharmacopoeia limits and preventing batch rejection.
Q: What represents the primary challenge in isolating this specific impurity?
A: The primary challenge lies in the structural similarity to the starting material and other byproducts. The patented method overcomes this by optimizing the Grignard reaction conditions and utilizing specific gradient elution chromatography to achieve a purity of 99.6%.
Q: How does this reference standard support regulatory filings?
A: Regulatory agencies like the FDA and EMA require that all impurities above the identification threshold (typically 0.10%) be characterized and controlled. This standard provides the necessary benchmark for validation of analytical methods, facilitating smoother New Drug Application (NDA) approvals.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Etoricoxib Impurity Supplier
At NINGBO INNO PHARMCHEM, we understand that the complexity of modern drug synthesis demands equally sophisticated quality control solutions. Our expertise extends beyond simple catalog supply; we specialize in the custom synthesis and isolation of challenging pharmaceutical impurities and reference standards. Leveraging our extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, we apply the same rigor to milligram-scale reference standard production. Our state-of-the-art facilities are equipped with rigorous QC labs capable of delivering the stringent purity specifications required by global pharmacopoeias, ensuring that every vial of Etoricoxib Impurity I we supply is accompanied by comprehensive analytical data, including NMR, MS, and HPLC profiles.
We invite procurement leaders and R&D directors to collaborate with us to secure their supply chains against regulatory uncertainty. By partnering with our technical team, you gain access to a Customized Cost-Saving Analysis that evaluates your current impurity control strategies against our advanced solutions. We encourage you to contact our technical procurement team today to request specific COA data for Compound I and to discuss route feasibility assessments for your unique manufacturing challenges. Let us help you turn quality control from a bottleneck into a competitive advantage.