Advanced Synthesis of Omacycline Intermediates: Overcoming Impurity Challenges for Commercial Scale
The pharmaceutical industry is constantly seeking robust synthetic routes for next-generation tetracycline antibiotics, particularly for advanced candidates like omacycline. A recent technological breakthrough detailed in patent CN114591216A introduces a highly efficient method for synthesizing key omacycline intermediates, specifically targeting the critical M1 and M2 precursors. This innovation addresses long-standing challenges in regioselectivity and byproduct management that have historically plagued the manufacturing of complex tetracycline derivatives. By optimizing the acid catalysis system and solvent conditions, this new methodology not only enhances the chemical purity of the intermediates but also offers significant advantages in terms of process economics and operational throughput. For global supply chain leaders and R&D directors, understanding these mechanistic improvements is essential for securing a reliable source of high-quality active pharmaceutical ingredients.
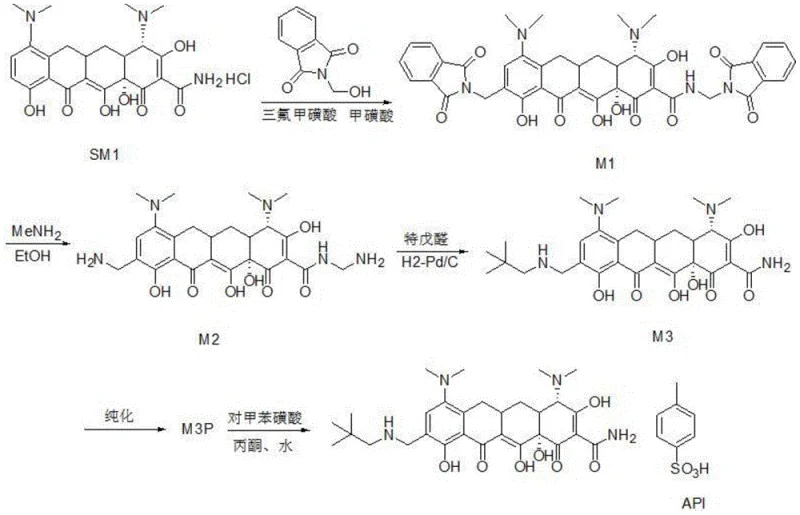
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
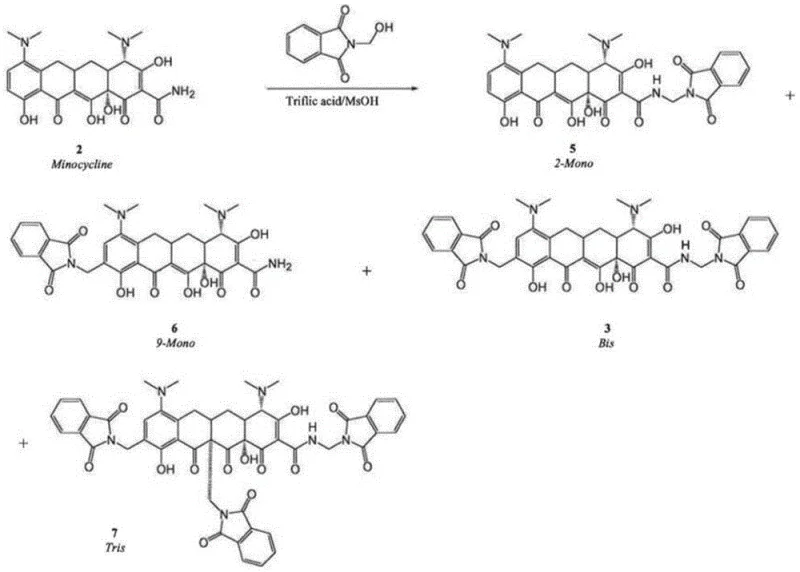
Traditional synthetic pathways for omacycline intermediates often suffer from poor regiocontrol during the substitution reactions on the tetracycline core. In the synthesis of the M1 intermediate, conventional acid-catalyzed methods frequently generate a complex mixture of impurities, including 2-position mono-substituted (2-Mono), 9-position mono-substituted (9-Mono), and trisubstituted (Tris) byproducts. The presence of the 2-Mono impurity is particularly detrimental because it cannot be converted into the desired downstream product, effectively becoming a permanent waste stream that lowers overall yield and complicates purification. Furthermore, while 9-Mono and Tris impurities can theoretically be converted in later steps, their presence necessitates prolonged reaction times and harsher conditions, which increases energy consumption and the risk of degrading the sensitive tetracycline scaffold. Similarly, in the subsequent conversion to M2, older protocols using ethanol solvents tend to produce significant amounts of the M2B byproduct, which reacts sluggishly in the following step, creating a bottleneck that extends the total production cycle time significantly.
The Novel Approach
The patented methodology overcomes these deficiencies through a dual-strategy optimization of both the catalytic environment and the reaction medium. For the preparation of M1, the inventors discovered that replacing single-acid systems with a specific binary mixture of trifluoromethanesulfonic acid and trifluoroacetic acid creates a unique reactivity profile. By maintaining a mass ratio of trifluoromethanesulfonic acid to trifluoroacetic acid between 3:7 and 3:2, the reaction selectively favors the desired substitution pattern while kinetically suppressing the formation of the problematic 2-Mono, 9-Mono, and Tris impurities. This results in a dramatic improvement in crude purity, with the main M1 peak reaching up to 84.56% yield while impurity levels are minimized. In the subsequent step to generate M2, the process switches to a methanol-based solvent system supplemented with 5-10% triethylamine. This modification effectively inhibits the formation of the M2B byproduct, ensuring that the reaction proceeds rapidly to the desired amine product with yields approaching 100%, thereby streamlining the entire synthetic sequence.
Mechanistic Insights into Mixed-Acid Catalyzed Substitution
The core innovation in the M1 synthesis lies in the nuanced interaction between the strong sulfonic acid and the carboxylic acid within the reaction matrix. Trifluoromethanesulfonic acid acts as a potent proton donor to activate the electrophilic centers on the tetracycline ring, facilitating the nucleophilic attack by N-hydroxymethylphthalimide. However, using it alone or with weaker acids like methanesulfonic acid often leads to over-activation, causing non-selective attacks at the C2 and C9 positions. The introduction of trifluoroacetic acid appears to modulate the acidity and solvation shell around the reactive intermediates, likely through hydrogen bonding networks that stabilize the transition state leading to the desired C7 (or specific target) substitution while destabilizing the transition states for the unwanted isomers. This fine-tuning of the acid strength and steric environment allows for a much cleaner reaction profile, where the thermodynamic drive towards the stable main product outcompetes the kinetic formation of side products. Consequently, the burden on downstream purification is vastly reduced, as the crude material contains significantly fewer structural analogs that are notoriously difficult to separate via crystallization.
In the transformation of M1 to M2, the mechanism shifts to an aminolysis reaction where the phthalimide protecting groups are removed and replaced with amine functionalities. The persistent issue in prior art was the incomplete conversion or side-reactions leading to the M2B species, which possesses a different substitution pattern that is kinetically inert in the subsequent conversion to the free base M3. The addition of triethylamine in the methanol solvent serves a dual purpose: it acts as a base to neutralize generated acids and potentially coordinates with the intermediate species to prevent the rearrangement or alternative attack pathways that lead to M2B. By ensuring that the reaction environment remains slightly basic yet nucleophilic due to the methylamine, the process drives the equilibrium decisively towards the M2 product. This mechanistic control is crucial because it eliminates the "tail" of slow-reacting material that would otherwise require extended heating, which could degrade the sensitive tetracycline core, thus preserving the integrity of the final API precursor.
How to Synthesize Omacycline Intermediate Efficiently
The implementation of this synthesis route requires precise control over reaction parameters, particularly temperature and acid ratios, to replicate the high yields reported in the patent data. The process begins with the careful preparation of the mixed acid system under nitrogen protection to prevent moisture interference, followed by the batch-wise addition of minocycline hydrochloride to manage exotherms. Detailed standard operating procedures regarding the specific addition rates, vacuum degassing steps to remove acid gases, and the precise quenching protocols into ice-water are critical for success. The subsequent isolation of the M1 salt involves pH adjustment and solvent swapping, which must be executed with high fidelity to ensure the correct crystal form and purity are obtained before moving to the aminolysis step. For a comprehensive breakdown of the exact temperatures, addition times, and workup procedures required to achieve these results, please refer to the standardized guide below.
- Prepare M1 by reacting minocycline hydrochloride with N-hydroxymethylphthalimide using a specific mass ratio of trifluoromethanesulfonic acid to trifluoroacetic acid (3: 7 to 3:2) to suppress regio-isomeric impurities.
- Isolate the M1 salt via aqueous quenching and pH adjustment, ensuring high purity before proceeding to the next step.
- Convert M1 to M2 using methylamine in methanol with 5-10% triethylamine additive to inhibit the formation of the slow-reacting M2B byproduct.
Commercial Advantages for Procurement and Supply Chain Teams
From a commercial perspective, the adoption of this patented synthesis route offers profound benefits for procurement managers and supply chain directors focused on cost efficiency and reliability. The primary advantage stems from the substantial reduction in raw material waste. By effectively suppressing the formation of the 2-Mono impurity, which is unrecoverable in traditional processes, the new method ensures that a much higher percentage of the expensive minocycline starting material is converted into valuable product. This directly translates to a lower cost of goods sold (COGS) per kilogram of intermediate, as less feedstock is required to produce the same output. Furthermore, the minimization of other impurities reduces the need for extensive recrystallization or chromatographic purification steps, which are often the most costly and time-consuming parts of pharmaceutical manufacturing. The elimination of these purification bottlenecks not only saves on solvent and labor costs but also accelerates the overall batch cycle time, allowing manufacturing facilities to increase their annual production capacity without requiring new capital equipment investments.
- Cost Reduction in Manufacturing: The shift to a mixed-acid system and optimized solvent conditions eliminates the generation of hard-to-remove impurities, thereby removing the need for expensive and yield-loss-inducing purification steps. This streamlined workflow significantly lowers the consumption of solvents and reagents while maximizing the utilization of high-value starting materials, resulting in a more economically viable production process for large-scale API manufacturing.
- Enhanced Supply Chain Reliability: By shortening the reaction times for both the M1 and M2 synthesis steps, the overall lead time for producing the intermediate is drastically reduced. Faster cycle times mean that manufacturers can respond more agilely to fluctuations in market demand, reducing the risk of stockouts. Additionally, the higher consistency in product quality reduces the likelihood of batch failures or out-of-specification results, ensuring a steady and predictable flow of materials to downstream API synthesis sites.
- Scalability and Environmental Compliance: The process utilizes common industrial solvents such as methanol, acetone, and trifluoroacetic acid, which are readily available and easier to recover and recycle compared to exotic reagents. The reduction in side products also means a lower load on waste treatment facilities, as there is less hazardous chemical waste to dispose of per unit of product. This aligns well with modern green chemistry initiatives and simplifies the regulatory compliance burden associated with environmental discharge, making the process highly scalable from pilot plant to commercial tonnage production.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this omacycline intermediate synthesis technology. These answers are derived directly from the experimental data and beneficial effects described in the patent documentation, providing clarity on how this method compares to legacy processes. Understanding these specifics is vital for technical teams evaluating the feasibility of integrating this route into their existing manufacturing portfolios.
Q: How does the new acid system improve the purity of the M1 intermediate?
A: The patented method utilizes a precise mixture of trifluoromethanesulfonic acid and trifluoroacetic acid. This specific acidic environment effectively inhibits the formation of difficult-to-remove 2-position and 9-position mono-substituted impurities, as well as trisubstituted byproducts, raising the main peak purity significantly compared to conventional single-acid methods.
Q: Why is the reduction of M2B impurity critical for production efficiency?
A: In traditional processes, the M2B byproduct converts very slowly in subsequent steps, acting as a bottleneck that prolongs reaction times and increases energy consumption. The new methanol/triethylamine solvent system drastically reduces M2B generation, thereby accelerating the overall throughput and reducing cycle times.
Q: Can this synthesis route be scaled for commercial API production?
A: Yes, the process is designed for scalability. By eliminating complex purification steps required to remove structural isomers and utilizing standard solvents like methanol and acetone, the route offers a robust pathway for manufacturing high-purity omacycline intermediates on a multi-ton scale.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Omacycline Intermediate Supplier
As the global demand for advanced tetracycline antibiotics continues to grow, securing a supply partner with deep technical expertise in complex intermediate synthesis is paramount. NINGBO INNO PHARMCHEM stands at the forefront of this sector, leveraging cutting-edge process chemistry to deliver high-purity pharmaceutical intermediates. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that we can meet the rigorous volume requirements of multinational pharmaceutical companies. We operate with stringent purity specifications and maintain rigorous QC labs equipped with state-of-the-art analytical instrumentation to guarantee that every batch of omacycline intermediate meets the highest international standards for safety and efficacy.
We invite procurement leaders and R&D directors to engage with us to explore how our optimized synthesis capabilities can enhance your supply chain resilience. By partnering with NINGBO INNO PHARMCHEM, you gain access to a Customized Cost-Saving Analysis tailored to your specific volume needs and quality targets. We encourage you to contact our technical procurement team today to request specific COA data and route feasibility assessments, allowing us to demonstrate how our advanced manufacturing protocols can drive value and efficiency in your omacycline development programs.