Scalable Synthesis of 2,6-Dimethyl-Tyrosine for Advanced Peptide Drug Development
Introduction to Advanced 2,6-Dimethyl-Tyrosine Manufacturing
The pharmaceutical industry's relentless pursuit of potent opioid analgesics has placed unnatural amino acids like 2,6-dimethyl-tyrosine at the forefront of peptide drug design. A pivotal advancement in this domain is detailed in patent CN108383744B, which discloses a robust and economically viable synthesis method for this critical building block. Unlike traditional approaches that rely on scarce resources, this novel methodology leverages a strategic three-step sequence starting from readily available phenolic precursors. The process is characterized by its operational simplicity, exceptional stability, and high overall yields, addressing the long-standing challenges of cost and scalability in fine chemical manufacturing. By shifting away from complex asymmetric hydrogenation, this technology offers a reliable pathway for producing high-purity intermediates essential for next-generation therapeutic agents.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of 2,6-dimethyl-tyrosine has been plagued by significant economic and technical barriers that hinder widespread adoption in commercial supply chains. Prior art literature frequently describes routes dependent on asymmetric hydrogenation using expensive noble metal catalysts, such as rhodium complexes with chiral diphosphine ligands. These methods not only incur prohibitive raw material costs but also introduce severe purification challenges related to residual heavy metal removal, which is strictly regulated in pharmaceutical applications. Furthermore, alternative strategies involving chiral Schiff base nickel complexes often suffer from multi-step sequences with low stereoselectivity and poor overall yields. These conventional pathways create bottlenecks in production capacity, leading to extended lead times and inconsistent supply availability for downstream peptide synthesizers who require bulk quantities of this specialized amino acid.
The Novel Approach
In stark contrast, the innovative route outlined in the patent data presents a streamlined solution that bypasses the need for precious metals entirely. The core of this breakthrough lies in a highly efficient chloromethylation followed by a condensation with acetamidomalonate, culminating in a straightforward hydrolysis. This approach utilizes inexpensive and abundant reagents such as chlorosulfonic acid, paraformaldehyde, and ferric trichloride, drastically lowering the entry barrier for production. The elimination of sensitive catalytic systems enhances process robustness, allowing for wider operating windows and reduced risk of batch failure. By adopting this chemistry, manufacturers can achieve a significant reduction in manufacturing costs while maintaining the stringent purity profiles required for clinical-grade materials.
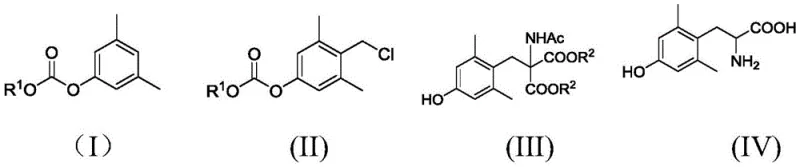
Mechanistic Insights into FeCl3-Catalyzed Chloromethylation and Condensation
The success of this synthetic strategy hinges on the precise control of electrophilic aromatic substitution during the initial chloromethylation step. In this transformation, the interaction between the phenolic substrate and the chloromethylating agent, generated in situ from chlorosulfonic acid and an aldehyde source, is mediated by a Lewis acid catalyst. Ferric trichloride (FeCl3) has been identified as the optimal catalyst, promoting the formation of the electrophilic chloromethyl cation which selectively attacks the para-position of the 3,5-dimethylphenol ring. The steric bulk of the methyl groups at the ortho-positions effectively blocks substitution at those sites, ensuring high regioselectivity for the desired 4-chloromethyl derivative. This selectivity is crucial for minimizing the formation of isomeric impurities that would be difficult to separate in later stages, thereby safeguarding the overall purity of the final amino acid product.
Following the formation of the chloromethyl intermediate, the synthesis proceeds via a nucleophilic substitution mechanism where the chloride leaving group is displaced by the enolate of diethyl acetamidomalonate. This reaction is facilitated by a strong base, typically sodium methoxide, which generates the reactive nucleophile under mild conditions ranging from 0°C to 50°C. The subsequent hydrolysis and decarboxylation step involves heating the protected malonate derivative in a strong acidic medium, such as hydrobromic or hydrochloric acid. This final transformation cleaves the ester and amide protecting groups while simultaneously removing the malonic acid moiety to reveal the free alpha-amino acid functionality. The rigorous control of pH during the workup, specifically adjusting to pH 5-6, ensures the precipitation of the zwitterionic product in high purity, effectively separating it from acidic byproducts and salts.
How to Synthesize 2,6-Dimethyl-Tyrosine Efficiently
Implementing this synthesis requires careful attention to reaction parameters to maximize yield and minimize impurity generation. The process begins with the solvent-free chloromethylation, which demands precise temperature control initially at -20°C to 0°C before warming to room temperature to drive the reaction to completion. Following isolation of the chloromethyl intermediate, the condensation with acetamidomalonate is performed in alcoholic solvents like methanol or ethanol, where the stoichiometry of the base is critical to prevent side reactions. The final hydrolysis step requires prolonged reflux times, typically between 3 to 5 hours, to ensure complete deprotection and decarboxylation. For a comprehensive guide on the specific molar ratios, workup procedures, and purification techniques validated by experimental data, please refer to the standardized protocol below.
- Perform chloromethylation of O-alkoxycarbonyl-3,5-dimethylphenol using chlorosulfonic acid and paraformaldehyde with FeCl3 catalyst under solvent-free conditions.
- React the resulting chloromethyl intermediate with diethyl acetamidomalonate in the presence of sodium methoxide in methanol to form the protected malonate derivative.
- Execute acidic hydrolysis and decarboxylation using hydrobromic or hydrochloric acid under reflux to yield the final 2,6-dimethyl-tyrosine product.
Commercial Advantages for Procurement and Supply Chain Teams
From a strategic sourcing perspective, this manufacturing route offers profound advantages that directly impact the bottom line and supply security for pharmaceutical companies. The primary driver of value is the complete elimination of noble metal catalysts, which removes a major variable cost component and mitigates the supply risk associated with volatile precious metal markets. Additionally, the use of commodity chemicals like formaldehyde sources and iron salts ensures that raw material procurement is stable and unaffected by geopolitical shortages that often plague specialty reagents. The process design inherently supports green chemistry principles, particularly through the solvent-free nature of the first step, which reduces waste disposal costs and environmental compliance burdens. These factors combine to create a supply chain that is not only more cost-effective but also more resilient against external disruptions.
- Cost Reduction in Manufacturing: The economic benefits of this route are derived from the substitution of high-cost catalytic systems with inexpensive Lewis acids and the simplification of the purification workflow. By avoiding the need for chromatographic removal of heavy metals, manufacturers can significantly reduce processing time and consumable costs associated with scavenger resins. The high yields reported in the patent examples, often exceeding 80% per step, further contribute to cost efficiency by maximizing the output from each unit of raw material input. This cumulative effect results in a substantially lower cost of goods sold, enabling more competitive pricing for the final API or peptide therapeutic.
- Enhanced Supply Chain Reliability: The reliance on widely available industrial chemicals rather than bespoke chiral ligands ensures a consistent and reliable supply of key inputs. Suppliers of chlorosulfonic acid and dimethylphenols are numerous globally, reducing the risk of single-source dependency. Furthermore, the robustness of the reaction conditions means that production can be maintained across different manufacturing sites without significant re-optimization, providing flexibility in case of regional disruptions. This stability is critical for long-term supply agreements where continuity of supply is paramount for clinical trial timelines and commercial launch schedules.
- Scalability and Environmental Compliance: The solvent-free condition employed in the initial chloromethylation step represents a significant engineering advantage for scale-up, as it eliminates the need for large solvent recovery systems and reduces the fire hazard profile of the process. The subsequent steps utilize common solvents like methanol and ethyl acetate, which are easily recycled and managed within standard waste treatment facilities. This alignment with environmental, health, and safety (EHS) standards facilitates faster regulatory approvals for new manufacturing sites. The ability to scale from kilogram to multi-ton quantities without fundamental changes to the chemistry ensures that the supply can grow in tandem with the commercial demand for the downstream drug product.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of 2,6-dimethyl-tyrosine synthesized via this method. These insights are derived directly from the experimental data and process descriptions found in the underlying patent literature. Understanding these details helps stakeholders evaluate the feasibility of integrating this intermediate into their specific development pipelines. The answers reflect the consensus on best practices for handling and processing this compound to ensure optimal quality.
Q: What are the advantages of this synthesis route over noble metal catalysis?
A: This route eliminates the need for expensive rhodium catalysts and chiral ligands, significantly reducing raw material costs and simplifying the purification process by avoiding heavy metal residues.
Q: How is regioselectivity controlled during the chloromethylation step?
A: The use of specific Lewis acid catalysts like ferric trichloride combined with steric hindrance from the 3,5-dimethyl groups ensures selective substitution at the 4-position of the phenol ring.
Q: Is this process suitable for large-scale commercial production?
A: Yes, the first step operates under solvent-free conditions which enhances safety and throughput, while the subsequent steps utilize common industrial solvents and reagents, facilitating easy scale-up.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 2,6-Dimethyl-Tyrosine Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that high-quality unnatural amino acids play in the development of advanced peptide therapeutics. Our technical team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that we can meet your volume requirements regardless of the project stage. We are committed to delivering products with stringent purity specifications, supported by our rigorous QC labs that employ state-of-the-art analytical methods to verify identity and assay. Our facility is equipped to handle the specific processing needs of this synthesis route, guaranteeing a consistent supply of 2,6-dimethyl-tyrosine that meets global regulatory standards.
We invite you to collaborate with us to optimize your supply chain for this essential intermediate. Contact our technical procurement team today to request a Customized Cost-Saving Analysis tailored to your specific volume needs. We are prepared to provide specific COA data from recent batches and conduct detailed route feasibility assessments to demonstrate how our manufacturing capabilities can support your long-term strategic goals. Let us be your partner in bringing innovative peptide drugs to market efficiently and reliably.