Advanced Continuous Flow Synthesis of 2-Fluoromalonic Diesters for Pharmaceutical Applications
The pharmaceutical and fine chemical industries are constantly seeking robust methodologies for introducing fluorine atoms into organic scaffolds, a critical modification that enhances metabolic stability and bioavailability in drug candidates. A significant technological breakthrough in this domain is detailed in Chinese Patent CN110437069B, which discloses a novel continuous synthesis method for 2-fluoro-malonic diester compounds. This patent addresses the longstanding challenges associated with producing these vital fluorine-containing intermediates by shifting from hazardous batch protocols to a sophisticated continuous flow decarbonylation process. By utilizing 2-fluoro-3-oxosuccinic acid diester as a raw material, the invention achieves the elimination of a carbonyl group under controlled high-temperature conditions within a continuous reactor system. This approach not only mitigates the severe safety risks inherent in handling reactive fluorinated species at elevated temperatures but also dramatically improves production efficiency and product purity, establishing a new benchmark for reliable pharmaceutical intermediate supplier capabilities in the fluorinated chemical sector.
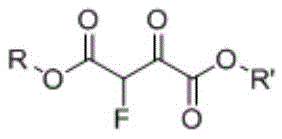
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the manufacturing of 2-fluoro-malonic diesters has been plagued by significant technical and safety hurdles that hinder cost reduction in API manufacturing. Traditional domestic methods often rely on hydrogen fluoride salt fluorination, a process notorious for its extreme toxicity and the strong corrosive nature of the reagents, which demands specialized, expensive equipment and generates substantial nitrogen-containing wastewater. Alternative routes described in prior art, such as condensation reactions involving fluoro ethyl acetate, suffer from abysmally low total yields, often hovering around 32.5%, alongside excessive three-waste generation. Furthermore, processes utilizing sodium hydride introduce severe explosion hazards due to hydrogen gas evolution, rendering them unsuitable for safe industrial mass production. Most critically, these legacy methods predominantly employ traditional kettle-type reactors, where the requirement for prolonged heating of large material volumes creates extensive high-temperature danger zones, leading to thermal decomposition of sensitive intermediates and inconsistent product quality.
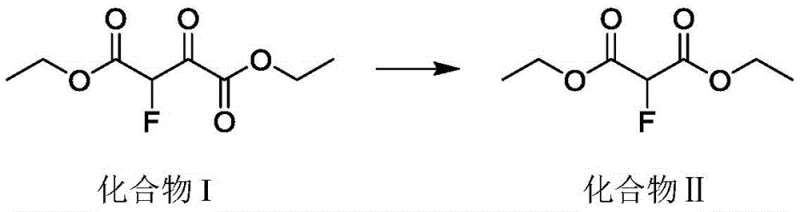
The Novel Approach
In stark contrast, the methodology outlined in CN110437069B leverages continuous reaction equipment to execute a high-temperature carbonyl elimination reaction with unprecedented control and safety. By transitioning to a tubular or column continuous reactor, the process ensures that the amount of material participating in the reaction per unit time is minimized, thereby shrinking the high-temperature dangerous area to a fraction of that found in batch systems. This flow chemistry approach allows the raw materials to be heated to the requisite reaction temperature of 200-500°C almost instantaneously, effectively avoiding the long intensification processes that cause raw material decomposition in kettles. The result is a streamlined synthesis where the yield is substantially improved, often exceeding 90%, while the automatic control system facilitates real-time feedback on temperature, pressure, and flow rate, ensuring consistent high-purity output suitable for commercial scale-up of complex esters.
Mechanistic Insights into Thermal Decarbonylation
The core chemical transformation driving this innovation is the thermal decarbonylation of the 3-oxo group in the 2-fluoro-3-oxosuccinic acid diester precursor. Under the specified high-temperature conditions, preferably between 250°C and 450°C, the molecule undergoes a concerted elimination of carbon monoxide. In a continuous flow environment, the heat transfer efficiency is vastly superior to that of batch vessels, allowing the reaction mixture to reach the activation energy threshold uniformly and rapidly. This precise thermal management is crucial because fluorinated beta-keto esters can be thermally sensitive; excessive residence time at peak temperatures in a static pot would lead to degradation, whereas the plug-flow nature of the reactor ensures that each molecular segment experiences the exact same thermal history. This uniformity is the key to suppressing side reactions and maximizing the conversion of the starting material into the desired 2-fluoro-malonic diester structure without compromising the integrity of the carbon-fluorine bond.
Furthermore, the continuous process offers distinct advantages regarding impurity control and downstream processing. In traditional batch fluorination, the presence of acidic byproducts and unreacted halides often necessitates complex neutralization and extraction steps that generate voluminous aqueous waste. However, the decarbonylation mechanism described here is essentially a self-reaction of the raw materials, potentially operable even under solvent-free conditions. When solvents are employed, the system allows for the use of stable, non-reactive options like diphenyl ether or high-boiling alkanes which do not interfere with the reaction pathway. The effluent from the continuous reactor contains primarily the target product and gaseous carbon monoxide, which can be easily vented or scrubbed. This simplicity allows for direct rectification of the crude product, achieving gas phase purities of over 98% with minimal post-treatment, thereby reducing lead time for high-purity intermediates and simplifying the overall manufacturing workflow.
How to Synthesize 2-Fluoromalonic Diester Efficiently
- Prepare the precursor solution by dissolving 2-fluoro-3-oxosuccinic acid diester in a suitable high-boiling solvent or use it neat, ensuring a homogeneous feed for the pump.
- Pump the solution continuously into a tubular or coil reactor maintained at a high temperature between 200°C and 500°C, with a system pressure of 0.5 to 10 MPa to maintain liquid phase.
- Collect the effluent and perform fractional distillation or rectification to separate the target 2-fluoromalonic diester from unreacted materials and byproducts, achieving purity over 98%.
To implement this advanced synthesis route effectively, manufacturers must adhere to strict operational parameters regarding flow rates and thermal gradients. The process begins with the preparation of the feed solution, where the 2-fluoro-3-oxosuccinic acid diester is either used neat or dissolved in a compatible high-boiling solvent to ensure smooth pumping. This solution is then continuously introduced into the heated reactor zone, where the residence time is tightly controlled between 5 to 50 minutes, with a preferred window of 10 to 30 minutes to balance conversion and throughput. The system pressure must be maintained between 0.5 and 10 MPa to keep the reaction mixture in the liquid phase at these elevated temperatures, preventing vaporization which could disrupt flow dynamics. For a comprehensive understanding of the specific equipment setup and parameter optimization, the detailed standardized synthesis steps are provided in the guide below.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this continuous flow technology represents a paradigm shift in sourcing strategies for fluorinated building blocks. The transition from batch to continuous processing fundamentally alters the cost structure and reliability profile of the supply chain, addressing critical pain points related to safety compliance, waste disposal, and production consistency. By eliminating the need for highly toxic hydrogen fluoride salts and corrosive reagents, the process removes significant liability and environmental remediation costs from the manufacturing equation. Moreover, the ability to operate the reactor continuously for extended periods, potentially 24/7 with standby equipment, ensures a steady stream of product that is not subject to the stop-start cycles of batch manufacturing, thereby enhancing supply chain reliability and reducing the risk of stockouts for downstream API producers.
- Cost Reduction in Manufacturing: The economic benefits of this process are driven by the drastic simplification of the reaction workflow and the elimination of expensive auxiliary reagents. By avoiding the use of triethylamine hydrogen fluoride salts and sodium hydride, the raw material costs are significantly lowered, and the expenses associated with hazardous waste treatment are virtually eliminated. Additionally, the solvent-free option or the use of recoverable high-boiling solvents reduces the consumption of volatile organic compounds, leading to substantial cost savings in both material procurement and environmental compliance. The high yield achieved through precise thermal control further amplifies these savings by maximizing the output per unit of input, ensuring that the cost reduction in pharmaceutical intermediate manufacturing is both immediate and sustainable.
- Enhanced Supply Chain Reliability: The continuous nature of the reaction equipment inherently supports a more robust supply chain model. Unlike batch processes that are limited by vessel size and require lengthy cooling and cleaning cycles between runs, the flow reactor can maintain production indefinitely as long as feedstock is available. This capability allows suppliers to respond more agilely to fluctuations in market demand, reducing lead times and ensuring consistent availability of high-purity materials. The reduced safety risk also means fewer unplanned shutdowns due to safety incidents or regulatory inspections, providing buyers with a more predictable and secure source of critical fluorinated intermediates for their drug development pipelines.
- Scalability and Environmental Compliance: Scaling this process does not require the construction of massive, high-risk pressure vessels; instead, capacity can be increased by extending the duration of the run or by numbering up identical reactor modules. This modularity facilitates a smoother transition from pilot-scale validation to full commercial production, minimizing the capital expenditure required for capacity expansion. From an environmental perspective, the process aligns with green chemistry principles by minimizing waste generation and avoiding the release of toxic fluorine-containing byproducts. The simplified post-treatment, often requiring only rectification, reduces the energy footprint of the separation process, making it an environmentally compliant choice for modern chemical manufacturing facilities aiming to reduce their carbon footprint.
A: The continuous method significantly reduces the inventory of hazardous materials at high temperatures at any given moment. Unlike traditional kettle reactions which require heating large volumes for extended periods, the flow reactor minimizes the 'high-temperature danger zone,' drastically lowering the risk of thermal runaway and equipment corrosion associated with older fluorination methods. A: Yes, the patent explicitly describes that the decarbonylation reaction can be carried out under solvent-free conditions. This capability eliminates the need for solvent recovery steps, reduces waste generation, and simplifies the downstream purification process, leading to a more environmentally friendly and cost-effective manufacturing route. A: The continuous flow architecture allows for precise control over residence time and temperature, preventing the decomposition often seen in batch heating. This results in yields exceeding 90% compared to roughly 40% in batch comparisons. Scalability is achieved not by building larger vessels, but by extending run times or numbering up reactor units, facilitating seamless transition from pilot to commercial tonnage.Frequently Asked Questions (FAQ)
Q: What are the safety advantages of the continuous decarbonylation method over traditional batch processes?
Q: Can this synthesis process be operated without solvents?
Q: How does the continuous process impact production scalability and yield?
The following questions address common technical and commercial inquiries regarding the continuous synthesis of 2-fluoromalonic diesters, based on the specific embodiments and comparative data provided in the patent literature. These insights are designed to clarify the operational feasibility and strategic advantages of adopting this flow chemistry platform for large-scale production. Understanding these nuances is essential for technical teams evaluating the integration of this technology into their existing manufacturing portfolios.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 2-Fluoromalonic Diester Supplier
At NINGBO INNO PHARMCHEM, we recognize the transformative potential of continuous flow chemistry in delivering high-value fluorinated intermediates to the global market. As a dedicated CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the theoretical benefits of patents like CN110437069B are fully realized in practical, industrial settings. Our facilities are equipped with state-of-the-art continuous flow reactors and rigorous QC labs capable of maintaining stringent purity specifications, guaranteeing that every batch of 2-fluoromalonic diester meets the exacting standards required for pharmaceutical applications. We are committed to bridging the gap between innovative academic research and reliable commercial supply.
We invite forward-thinking pharmaceutical companies and chemical manufacturers to collaborate with us to leverage this advanced synthesis technology. By partnering with our technical procurement team, you can request a Customized Cost-Saving Analysis tailored to your specific volume requirements and quality needs. We encourage you to reach out today to obtain specific COA data and route feasibility assessments, allowing us to demonstrate how our continuous manufacturing capabilities can optimize your supply chain and accelerate your drug development timelines.