Scalable Synthesis of Indazole Cyclotriazole Compounds for Next-Gen Kinase Inhibitors
Scalable Synthesis of Indazole Cyclotriazole Compounds for Next-Gen Kinase Inhibitors
The pharmaceutical landscape for Chronic Myelogenous Leukemia (CML) treatment is constantly evolving, driven by the urgent need to overcome drug resistance mechanisms such as the T315I mutation. Patent CN114573567B introduces a novel class of indazole cyclotriazole compounds that demonstrate potent inhibitory activity against Bcr-Abl tyrosine kinase. This technology leverages a fragment-based drug design strategy, integrating an indazole hinge-binding motif with a flexible L-proline linker and a terminal halogenated benzene ring. For R&D directors and procurement specialists in the oncology sector, this patent represents a significant opportunity to access high-purity pharmaceutical intermediates with a robust and scalable synthetic pathway. The structural versatility of this scaffold allows for the rapid generation of compound libraries, facilitating the identification of lead candidates with optimized pharmacokinetic profiles.
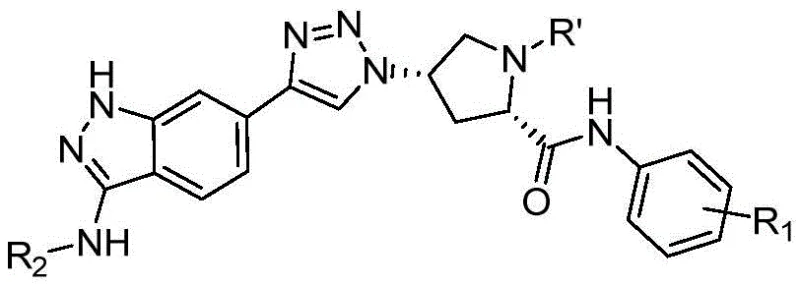
From a commercial manufacturing perspective, the synthesis outlined in this patent utilizes well-established organic transformations that are amenable to kilogram-scale production. The integration of click chemistry and Sonogashira coupling ensures high reaction efficiency while maintaining strict control over stereochemistry and impurity profiles. As a reliable pharmaceutical intermediate supplier, understanding the nuances of this synthetic route is critical for ensuring consistent quality and supply continuity for downstream API manufacturing. The following analysis details the technical advantages, mechanistic insights, and commercial viability of producing these advanced kinase inhibitor intermediates.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditional approaches to synthesizing tyrosine kinase inhibitors often rely on complex heterocyclic formations that require harsh reaction conditions, such as high temperatures and strong acids, which can compromise the integrity of sensitive functional groups. Furthermore, conventional methods frequently struggle with regioselectivity issues, leading to difficult-to-separate isomeric byproducts that necessitate extensive chromatographic purification, thereby driving up costs and reducing overall yield. Many existing Bcr-Abl inhibitors also face challenges related to the T315I gatekeeper mutation, rendering them ineffective in resistant patient populations. The synthetic routes for older generation inhibitors often involve multiple protection and deprotection steps that add unnecessary complexity and waste to the manufacturing process, making them less attractive for cost-sensitive commercial production.
The Novel Approach
The methodology described in patent CN114573567B overcomes these hurdles by employing a modular synthesis strategy centered around copper-catalyzed azide-alkyne cycloaddition (CuAAC), commonly known as click chemistry. This approach allows for the efficient coupling of the indazole core with the chiral proline linker under mild, aqueous-compatible conditions, significantly enhancing atom economy. By introducing halogenated substituents on the terminal benzene ring, the process enables fine-tuning of the electronic and steric properties of the molecule to maximize binding affinity against both wild-type and mutant kinases. This novel route eliminates the need for extreme reaction parameters, reduces the formation of hazardous byproducts, and streamlines the purification workflow, resulting in a more sustainable and economically viable manufacturing process for high-value oncology intermediates.
Mechanistic Insights into CuAAC and Sonogashira Coupling
The core of this synthetic strategy relies on two pivotal catalytic cycles: the Sonogashira cross-coupling and the Copper(I)-catalyzed Azide-Alkyne Cycloaddition. The synthesis begins with the formation of the indazole core via nucleophilic substitution of 4-bromo-2-fluorobenzonitrile with hydrazine, followed by a palladium-copper catalyzed Sonogashira reaction to install the terminal alkyne. This step is critical for establishing the conjugated system required for subsequent triazole formation. The mechanism involves the oxidative addition of the aryl halide to the Pd(0) catalyst, followed by transmetallation with the copper-acetylide species and reductive elimination to forge the carbon-carbon bond. Strict control of oxygen levels is essential here to prevent homocoupling of the alkyne, ensuring high purity of the ethynyl-indazole intermediate.
Subsequently, the chiral linker is prepared from L-hydroxyproline through Boc protection, mesylation, and azide substitution, preserving the stereochemical integrity essential for biological activity. The convergence of these two fragments occurs via the click chemistry reaction, where the terminal alkyne and the organic azide undergo a [3+2] cycloaddition catalyzed by Cu(I) generated in situ from copper sulfate and sodium ascorbate. This reaction proceeds through a copper-acetylide intermediate that coordinates with the azide, lowering the activation energy and directing the formation of the 1,4-disubstituted 1,2,3-triazole exclusively. This high regioselectivity is a major advantage for impurity control, as it prevents the formation of the 1,5-isomer, thereby simplifying downstream processing and ensuring the final product meets stringent pharmaceutical specifications.
How to Synthesize Indazole Cyclotriazole Efficiently
The preparation of these bioactive compounds follows a logical, step-wise progression that balances chemical efficiency with operational simplicity. The process initiates with the construction of the heterocyclic core, followed by the preparation of the chiral amino acid-derived linker, and culminates in their convergence via click chemistry. Each step has been optimized to utilize common laboratory solvents such as dichloromethane, tetrahydrofuran, and ethanol, which facilitates easy technology transfer from pilot plant to commercial scale. The use of Boc protecting groups throughout the synthesis provides stability to the amine functionalities during harsh transformation steps, while allowing for clean removal in the final stages using trifluoroacetic acid. Detailed standardized operating procedures for each reaction stage are essential to maintain batch-to-batch consistency.
- Condense 4-bromo-2-fluorobenzonitrile with hydrazine monohydrate to form the indazole core, followed by Sonogashira coupling to introduce the ethynyl group.
- Protect L-hydroxyproline with Boc, activate with mesyl chloride, and substitute with sodium azide to generate the chiral azide linker.
- Perform copper-catalyzed click chemistry between the ethynyl-indazole and the azide-proline, followed by acylation and deprotection to yield the final compound.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this synthetic route offers tangible benefits in terms of cost structure and logistical reliability. The reliance on commodity chemicals like L-hydroxyproline and substituted anilines means that raw material sourcing is not dependent on obscure or single-source vendors, mitigating supply chain risks. Furthermore, the reaction conditions are generally mild, often proceeding at room temperature or moderate reflux, which reduces energy consumption and lowers the burden on HVAC and utility systems in the manufacturing facility. The high selectivity of the click chemistry step minimizes the generation of complex waste streams, aligning with modern environmental compliance standards and reducing the costs associated with waste disposal and solvent recovery.
- Cost Reduction in Manufacturing: The streamlined synthesis eliminates the need for expensive transition metal scavengers often required in traditional cross-coupling reactions, as the copper catalyst loading is minimal and easily removed during aqueous workup. Additionally, the high yields observed in the click chemistry and acylation steps reduce the amount of starting material required per kilogram of output, directly lowering the cost of goods sold (COGS). By avoiding cryogenic conditions and utilizing ambient pressure reactions, the process also reduces capital expenditure on specialized reactor equipment, making it highly suitable for multi-purpose manufacturing suites.
- Enhanced Supply Chain Reliability: The synthetic pathway is robust and tolerant to minor variations in reagent quality, which ensures consistent production output even when facing fluctuations in raw material grades. The intermediates generated, such as the Boc-protected azide and the ethynyl-indazole, are stable and can be stocked as strategic buffers to decouple production stages, thereby smoothing out workflow bottlenecks. This modularity allows for flexible manufacturing scheduling, enabling the supply chain team to respond rapidly to changes in demand without compromising on delivery timelines or product quality.
- Scalability and Environmental Compliance: The process utilizes solvents that are widely accepted in pharmaceutical manufacturing and have established recovery protocols, supporting a circular economy approach within the plant. The absence of highly toxic reagents or explosive intermediates enhances operational safety, reducing insurance premiums and regulatory scrutiny. The scalability of the Sonogashira and click reactions has been proven in various industrial contexts, ensuring that the transition from gram-scale R&D to ton-scale commercial production can be achieved with minimal process re-engineering, securing long-term supply continuity for partners.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of these indazole cyclotriazole derivatives. These insights are derived directly from the experimental data and process descriptions found in the patent literature, providing a transparent view of the technology's capabilities. Understanding these details helps stakeholders make informed decisions regarding licensing, procurement, and development strategies.
Q: What is the primary advantage of the indazole-triazole scaffold in Bcr-Abl inhibition?
A: The indazole-triazole scaffold acts as a hinge region binding fragment that significantly improves affinity for the ATP site of Abl kinase, showing potent activity against both wild-type and T315I mutant kinases.
Q: How does the click chemistry step impact the overall process efficiency?
A: The copper-catalyzed azide-alkyne cycloaddition (CuAAC) offers high atom economy and regioselectivity under mild aqueous conditions, drastically simplifying purification and reducing solvent waste compared to traditional heterocycle formation.
Q: Are the starting materials for this synthesis commercially viable for large-scale production?
A: Yes, key precursors such as 4-bromo-2-fluorobenzonitrile and L-hydroxyproline are commodity chemicals available from reliable global suppliers, ensuring supply chain continuity for commercial manufacturing.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Indazole Cyclotriazole Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that high-quality intermediates play in the development of life-saving oncology therapies. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project transitions smoothly from the laboratory bench to the global market. We adhere to stringent purity specifications and operate rigorous QC labs equipped with state-of-the-art analytical instrumentation to guarantee that every batch of indazole cyclotriazole intermediate meets the highest industry standards. Our commitment to technical excellence allows us to navigate complex synthetic challenges, delivering materials that accelerate your drug development timeline.
We invite you to collaborate with us to leverage this innovative synthetic technology for your next-generation kinase inhibitor programs. Contact our technical procurement team today to request a Customized Cost-Saving Analysis tailored to your specific volume requirements. We are ready to provide specific COA data and comprehensive route feasibility assessments to demonstrate how our manufacturing capabilities can optimize your supply chain and reduce overall project costs.